Key Takeaways
Thrombotic thrombocytopenic purpura is a rare, life-threatening blood disorder in which tiny clots form in small blood vessels throughout the body, driven by a missing or blocked enzyme called ADAMTS13 [1,4].
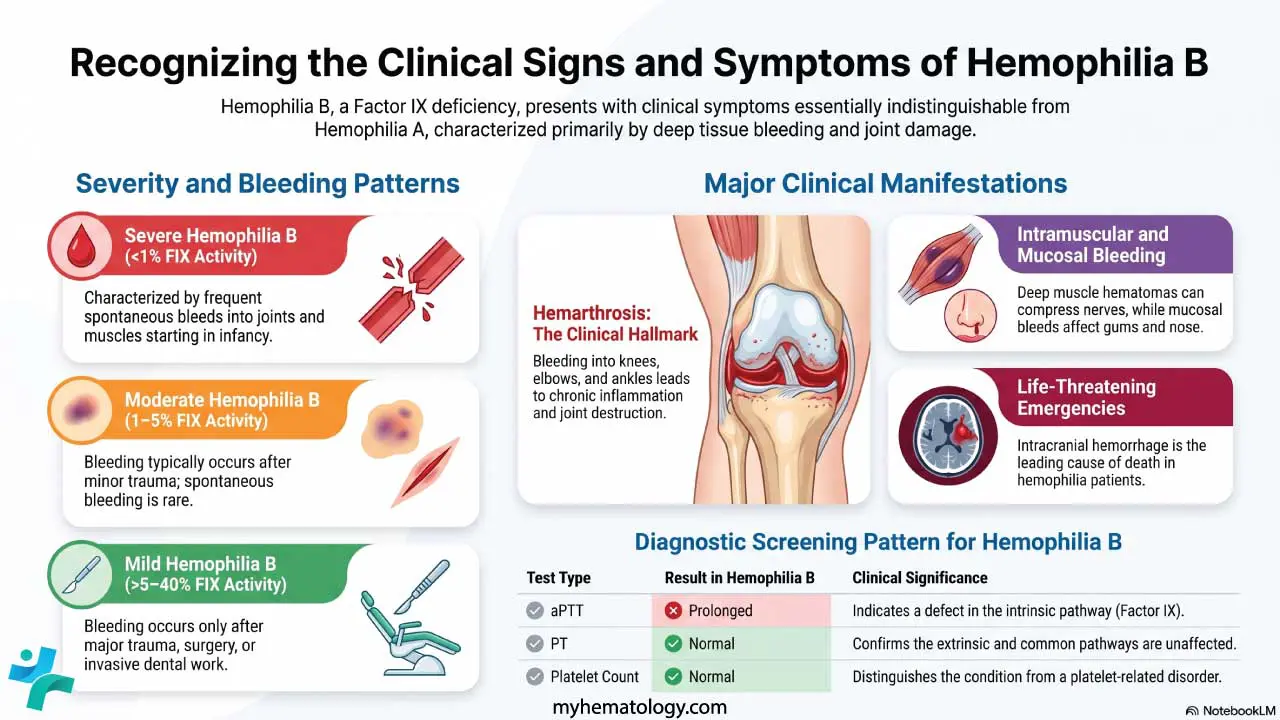
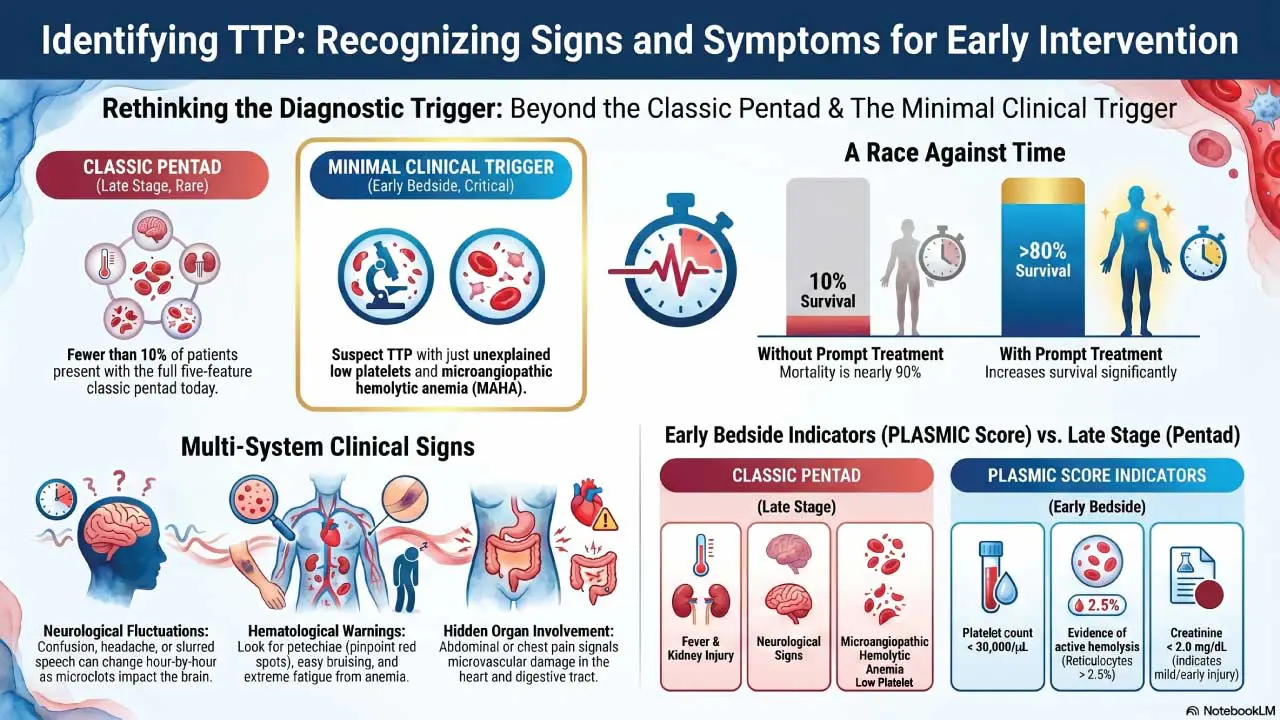
- Signs and symptoms ▾: The classic five-feature "pentad" (low platelets, anemia from red cell shredding, neurological signs, kidney injury, fever) appears in fewer than 10% of patients today; thrombocytopenia plus microangiopathic hemolytic anemia without another cause is enough to act on [1,7].
- Causes ▾: Inherited mutations or acquired causes like inhibitory IgG autoantibodies stimulated by infection, autoimmune/connective tissue disease, drugs, stem cell transplantation or cardiac surgery
- Pathophysiology ▾: TTP is primarily due to a deficiency or dysfunction of ADAMTS13. This deficiency leads to the formation of abnormally large VWF multimers, which can bind platelets excessively, triggering platelet aggregation and thrombus formation. These microthrombi can occlude small blood vessels, leading to tissue damage.
- Laboratory investigations ▾: Diagnosis combines clinical picture, schistocytes on a blood smear, normal coagulation tests, and severely reduced ADAMTS13 activity (below 10%). The PLASMIC score helps predict severe deficiency at the bedside [1,5].
- Treatment and management ▾: Frontline treatment for immune TTP combines plasma exchange, corticosteroids, caplacizumab, and early rituximab. A new recombinant ADAMTS13 product (apadamtase alfa, FDA-approved 2023) is now available for the inherited form [2,3,6].
*Click ▾ for more information
Introduction
Thrombotic thrombocytopenic purpura, usually shortened to TTP, sits at the dangerous intersection of clotting and bleeding. Patients form tiny clots in their smallest vessels while simultaneously running out of platelets to stop ordinary bleeding. The root cause is a single enzyme, ADAMTS13, that has either been silenced by autoantibodies or was never made properly because of a gene mutation [4,7].
This article walks through what thrombotic thrombocytopenic purpura is, how it damages the body, how clinicians diagnose it, and how treatment has evolved over the past decade.
Two forms of TTP
There are two forms, and they behave differently.
Immune (acquired) TTP is the common one, accounting for roughly 95% of cases. The immune system makes autoantibodies that block ADAMTS13. It typically strikes adults in their 30s and 40s, with women affected two to three times more often than men [8].
Congenital TTP, also called Upshaw–Schulman syndrome, comes from inherited mutations in the ADAMTS13 gene. It is rare, often presents in childhood or pregnancy, and is now treatable with regular plasma infusions or, more recently, recombinant ADAMTS13 [6].
Pathophysiology
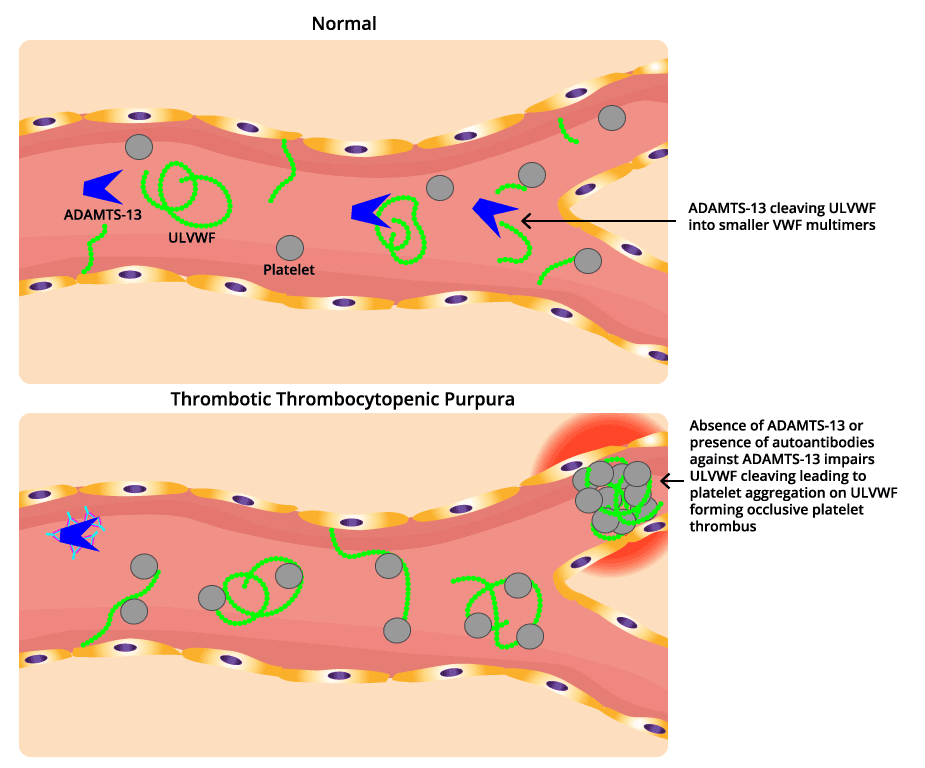
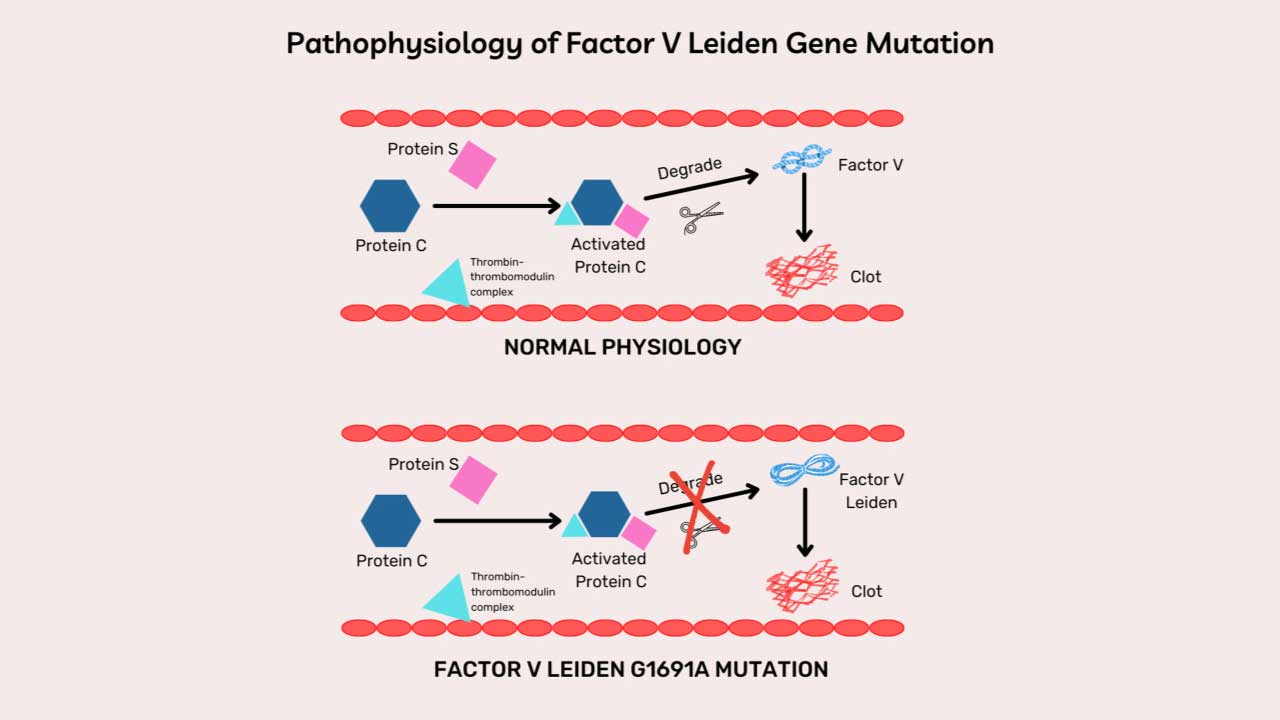
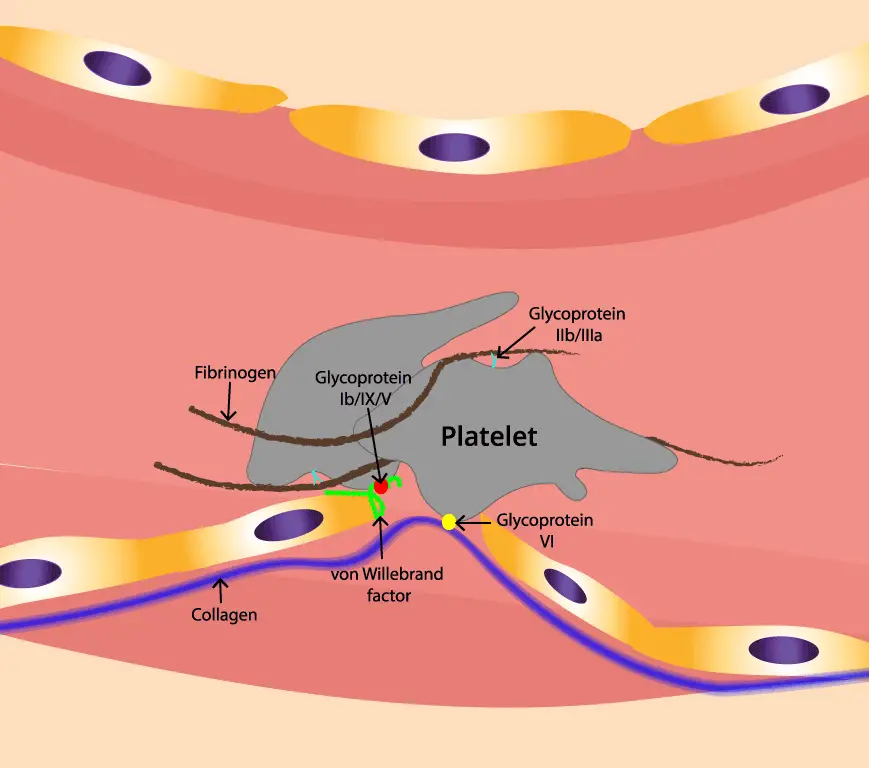
ADAMTS13, an enzyme responsible for cleaving large von Willebrand factor (VWF) multimers, plays a pivotal role in maintaining the delicate balance of blood vessel integrity. In thrombotic thrombocytopenic purpura (TTP), the absence of ADAMTS13 leads to an accumulation of these large VWF multimers, creating a sticky environment that promotes platelet aggregation.
To understand TTP, picture ADAMTS13 as a pair of molecular scissors patrolling the bloodstream. Its job is to trim long strings of von Willebrand factor (VWF) (a sticky protein that helps platelets plug damaged vessel walls) down to a working size [4].
When ADAMTS13 is missing or blocked, those VWF strings stay long. Long VWF strings act like flypaper for platelets, capturing them through the platelet GP1bα receptor and dragging them into clumps inside small blood vessels. Three things follow [1,4]:
- Microthrombi form in arterioles and capillaries, blocking blood flow to the brain, kidneys, heart, and other organs.
- Platelets get used up in these clots, dropping the platelet count and creating a paradoxical bleeding risk.
- Red blood cells are shredded as they squeeze through clot-narrowed vessels, producing schistocytes and microangiopathic hemolytic anemia (MAHA, or anemia caused by mechanical damage in small vessels).
In immune TTP, an IgG autoantibody binds and inhibits ADAMTS13. Triggers can include infections, pregnancy, certain drugs, autoimmune disease, stem cell transplant, or major surgery [4,7]. In congenital TTP, the gene that codes for ADAMTS13 is faulty, so the enzyme is never made in functional form [6].
When to Suspect TTP
Suspect TTP and start treatment when you see:
- Unexplained thrombocytopenia (low platelets), and
- Microangiopathic hemolytic anemia (anemia with schistocytes on the blood smear), and
- No better explanation (no DIC, no obvious sepsis driving it, no diarrheal illness pointing to HUS).
Rethinking the TTP Pentad
The classic pentad of low platelets, hemolytic anemia, neurological signs, kidney injury, and fever is now considered a late presentation. Waiting for all five costs lives [1,7].
Clinical Signs and Symptoms
Symptoms reflect where the microthrombi are landing.
Hematological. Easy bruising, petechiae (pinpoint red spots from tiny bleeds under the skin), nosebleeds, and fatigue or pallor from anemia.
Neurological. Headache, confusion, drowsiness, slurred speech, weakness, visual changes, or seizures. Neurological signs can fluctuate hour by hour.
Renal. Mild kidney impairment is common; severe kidney failure is more typical of hemolytic uremic syndrome than TTP.
General. Fever, malaise, abdominal pain, and chest pain (if cardiac microvessels are affected, TTP can cause myocardial damage that is easy to miss).
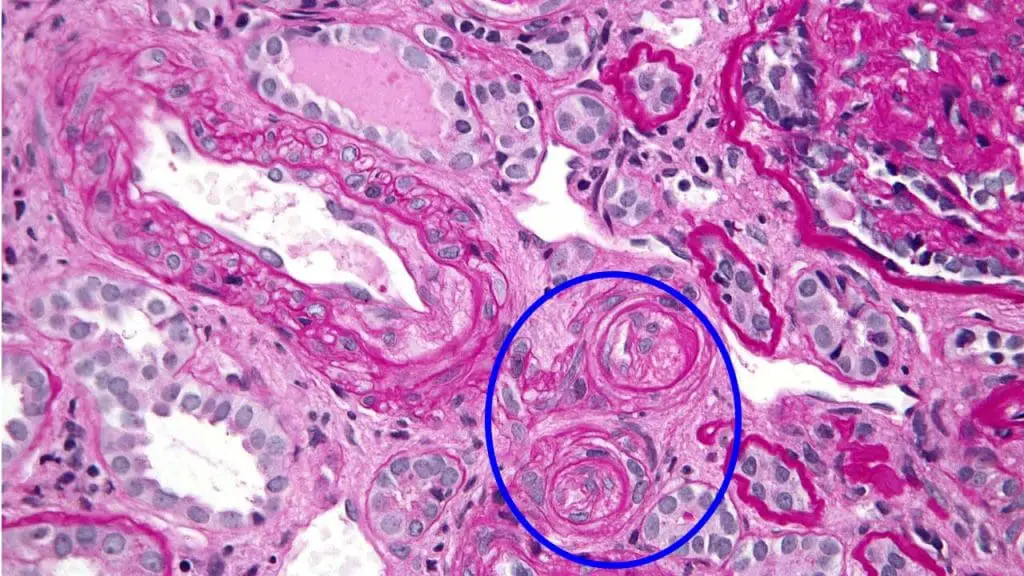
A kidney biopsy may show "onion skinning" (concentric layers of fibrin-like material in arteriole walls) but biopsy is rarely needed for diagnosis [1].
Laboratory Investigations of TTP
Diagnosis is built from common tests interpreted in context, with one specialized assay to confirm.
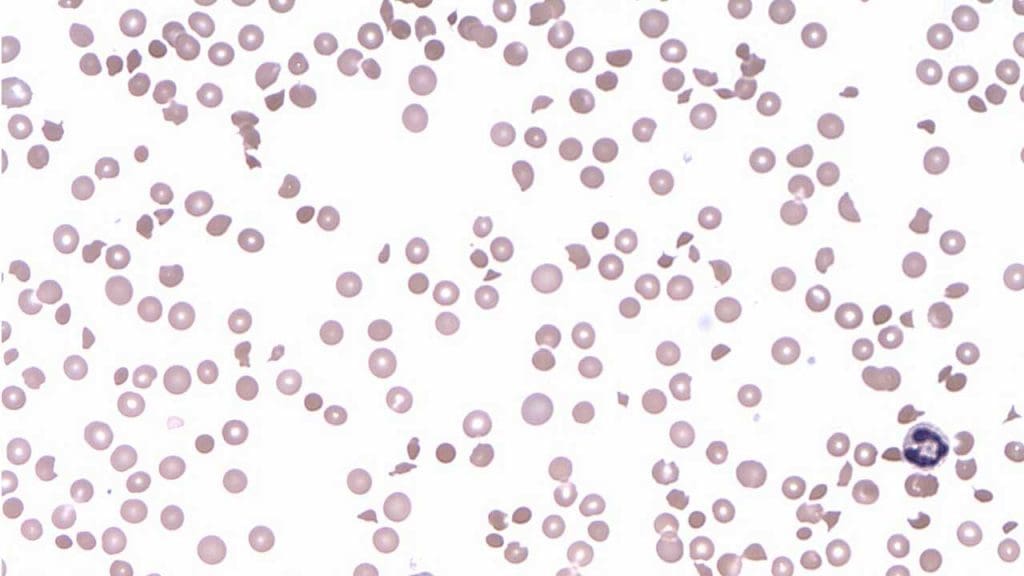
Complete blood count (CBC) and blood smear
- Severe thrombocytopenia, usually below 30,000/μL.
- Anemia with elevated reticulocytes, indicating the bone marrow is racing to replace destroyed red cells.
- Schistocytes on the peripheral blood smear — fragmented red cells produced by mechanical shearing. More than 1% schistocytes is a strong clue [1].
Markers of hemolysis
- Elevated lactate dehydrogenase (LDH).
- Decreased haptoglobin (consumed binding free hemoglobin).
- Elevated indirect bilirubin.
- Negative direct Coombs test, which separates TTP from autoimmune hemolytic anemia.
Coagulation studies
PT, aPTT, and fibrinogen are typically normal. This is one of the most useful distinctions from disseminated intravascular coagulation (DIC), where all three are deranged.
Renal panel
Mild creatinine elevation is common. Severe acute kidney injury suggests hemolytic uremic syndrome instead.
ADAMTS13 activity and inhibitor assay
The definitive test. ADAMTS13 activity below 10% confirms TTP. An anti-ADAMTS13 inhibitor antibody points to the immune (acquired) form; absence of an inhibitor with low activity in a young patient suggests the congenital form [1,4]. Treatment should never wait for this result.
PLASMIC score
The PLASMIC score is a seven-variable bedside tool that predicts severe ADAMTS13 deficiency before the assay returns [5]. The variables are:
- Platelet count below 30,000/μL (30 x 10⁹/L)
- Hemolysis evidence (reticulocyte count >2.5%, undetectable haptoglobin, or indirect bilirubin >2 mg/dL)
- No active cancer
- No history of solid organ or stem cell transplant
- MCV less than 90 fL
- INR less than 1.5
- Creatinine less than 2 mg/dL
A score of 6–7 indicates a high probability of TTP, supporting urgent plasma exchange.
PLASMIC Score for TTP
Predicts ADAMTS13 deficiency in suspected thrombotic thrombocytopenic purpura (TTP).
Differential Diagnosis of TTP
TTP is one of several thrombotic microangiopathies (TMAs) — disorders that damage small vessels through clot formation. Distinguishing them matters because treatments diverge sharply.
Treatment and Management
The goal is to restore working ADAMTS13, remove inhibitor antibodies, and stop platelet aggregation while these other steps take effect. Modern care for immune TTP rests on a triplet of therapies started together, with rituximab added early [2,3].
Plasma exchange (PEX)
Plasma exchange is the cornerstone. The patient's plasma that is carrying autoantibodies and uncleaved long VWF strings is removed and replaced with donor fresh frozen plasma, which also supplies functional ADAMTS13. PEX should start within hours of clinical suspicion, before the ADAMTS13 result is back [1,2]. Daily exchanges continue until the platelet count recovers and stabilizes.
Corticosteroids
Methylprednisolone or prednisone is started alongside PEX in immune TTP to suppress the autoantibody response and dampen inflammation [2].
Caplacizumab
Caplacizumab is a small antibody fragment that blocks the part of VWF that grabs platelets. Adding it to PEX and steroids speeds platelet recovery, shortens the time in hospital, and reduces TTP-related deaths. Based on the International Society on Thrombosis and Haemostasis (ISTH) guidelines, caplacizumab should be initiated early (ideally within 3 days of the first plasma exchange) for maximum benefit, rather than waiting to see if standard therapy fails [2,9]. However, because it prevents clotting, updated good practice statements advise that it should not be given alongside standard antithrombotic medications until a patient's platelet count safely recovers above 50,000/μL (50 x 10⁹/L) [9].
Rituximab
Rituximab depletes the B cells that make anti-ADAMTS13 antibodies. Once reserved for refractory or relapsing disease, current evidence supports adding it during the acute episode for immune TTP, where it shortens remission time and lowers relapse risk [2,4].
Recombinant ADAMTS13 for congenital TTP
For decades, patients with congenital TTP relied on frequent, burdensome fresh frozen plasma (FFP) infusions. Today, apadamtase alfa (Adzynma) (a recombinant ADAMTS13 enzyme replacement therapy) is the strongly recommended gold standard for cTTP prophylaxis. The 2025 ISTH focused update officially recommends using recombinant ADAMTS13 over plasma infusions for patients in remission, as it safely provides a targeted, sustained concentration of the enzyme, drastically reducing the risk of hidden long-term organ damage and clinical relapses [9].
Long-term management
Roughly 30–50% of immune TTP patients relapse, often within the first two years [4,8]. Standard follow-up includes:
- Regular monitoring of ADAMTS13 activity and inhibitor titers, even in remission.
- Preemptive rituximab if ADAMTS13 activity drops, before clinical relapse.
- Patient education on triggers (infections, pregnancy, certain drugs) and warning symptoms.
Frequently Asked Questions (FAQs)
Can you live a normal life with thrombotic thrombocytopenic purpura (TTP)?
Yes, most people who survive an acute episode of immune TTP return to normal daily life. Long-term care involves periodic blood tests to track ADAMTS13 activity and watching for triggers like infections or pregnancy. Some patients receive maintenance rituximab if their ADAMTS13 levels start to drop. With consistent follow-up, the outlook is far better than it was even a decade ago.
What are the triggers of thrombotic thrombocytopenic purpura?
Common triggers for immune TTP include infections (especially viral), pregnancy and the postpartum period, certain medications (quinine, ticlopidine, clopidogrel, gemcitabine, calcineurin inhibitors), autoimmune diseases such as lupus, major surgery (particularly cardiac), and stem cell transplantation. In many patients, no specific trigger is identified.
Can TTP be cured?
Acute episodes can be put into sustained remission with modern combination therapy, and many patients with immune TTP do not need lifelong drug therapy — only monitoring. Congenital TTP is not curable in the traditional sense but is now well controlled with regular plasma infusions or recombinant ADAMTS13 therapy. Calling TTP "incurable" is too pessimistic for most patients today.
What is the difference between TTP and immune thrombocytopenia (ITP)?
TTP and ITP both produce a low platelet count, but the mechanisms are different. TTP is a disorder of too much clotting in small blood vessels because ADAMTS13 cannot trim VWF, and platelets are consumed making microclots. ITP is an autoimmune disorder of platelet destruction: antibodies tag platelets for clearance by the spleen. TTP comes with anemia, schistocytes, and organ damage; ITP usually causes only bleeding symptoms with otherwise normal blood counts.
As a caregiver, what warning signs of TTP should send us to the emergency room?
Seek urgent care for new confusion, slurred speech, one-sided weakness, severe headache, seizures, unusual bruising or petechiae (pinpoint red spots), dark urine, or sudden pallor with fatigue. In a person already diagnosed with thrombotic thrombocytopenic purpura, any of these could signal a relapse and needs immediate evaluation.
How quickly does TTP need to be treated?
Within hours of clinical suspicion. Plasma exchange should begin the same day, before laboratory confirmation of ADAMTS13 deficiency is available. Untreated TTP has a mortality rate near 90%, while prompt treatment brings survival above 80% [4,7].
Glossary of Related Medical Terms
- Upshaw–Schulman syndrome: Inherited (congenital) TTP.
- ADAMTS13: A plasma enzyme that trims long strands of VWF.
- VWF (von Willebrand factor): A sticky protein that helps platelets plug damaged vessel walls.
- Microthrombus: A tiny blood clot in a small vessel.
- Thrombocytopenia: Low platelet count.
- MAHA: Anemia from red cells being shredded as they pass through clot-narrowed vessels.
- Schistocyte: A fragmented red blood cell on a smear.
- Plasma exchange (PEX): Removing a patient's plasma and replacing it with donor plasma.
- Caplacizumab: A drug that blocks VWF from grabbing platelets.
- Rituximab: An antibody drug that depletes B cells.
- Pentad: The five classical features of TTP, now considered a late presentation.
- PLASMIC score: A bedside tool predicting severe ADAMTS13 deficiency.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Zheng, X. L., Vesely, S. K., Cataland, S. R., Coppo, P., Geldziler, B., Iorio, A., Matsumoto, M., Mustafa, R. A., Pai, M., Rock, G., Russell, L., Tarawneh, R., Valdes, J., & Peyvandi, F. (2020). ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. Journal of thrombosis and haemostasis : JTH, 18(10), 2486–2495. https://doi.org/10.1111/jth.15006
- Zheng, X. L., Vesely, S. K., Cataland, S. R., Coppo, P., Geldziler, B., Iorio, A., Matsumoto, M., Mustafa, R. A., Pai, M., Rock, G., Russell, L., Tarawneh, R., Valdes, J., & Peyvandi, F. (2020). ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. Journal of thrombosis and haemostasis : JTH, 18(10), 2496–2502. https://doi.org/10.1111/jth.15010
- Scully, M., Cataland, S. R., Peyvandi, F., Coppo, P., Knöbl, P., Kremer Hovinga, J. A., Metjian, A., de la Rubia, J., Pavenski, K., Callewaert, F., Biswas, D., De Winter, H., Zeldin, R. K., & HERCULES Investigators (2019). Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine, 380(4), 335–346. https://doi.org/10.1056/NEJMoa1806311
- Sukumar, S., Lämmle, B., & Cataland, S. R. (2021). Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. Journal of clinical medicine, 10(3), 536. https://doi.org/10.3390/jcm10030536
- Bendapudi, P. K., Hurwitz, S., Fry, A., Marques, M. B., Waldo, S. W., Li, A., Sun, L., Upadhyay, V., Hamdan, A., Brunner, A. M., Gansner, J. M., Viswanathan, S., Kaufman, R. M., Uhl, L., Stowell, C. P., Dzik, W. H., & Makar, R. S. (2017). Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. The Lancet. Haematology, 4(4), e157–e164. https://doi.org/10.1016/S2352-3026(17)30026-1
- Scully, M., Knöbl, P., Kentouche, K., Rice, L., Windyga, J., Schneppenheim, R., Kremer Hovinga, J. A., Kajiwara, M., Fujimura, Y., Maggiore, C., Doralt, J., Hibbard, C., Martell, L., & Ewenstein, B. (2017). Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood, 130(19), 2055–2063. https://doi.org/10.1182/blood-2017-06-788026
- Joly, B. S., Coppo, P., & Veyradier, A. (2017). Thrombotic thrombocytopenic purpura. Blood, 129(21), 2836–2846. https://doi.org/10.1182/blood-2016-10-709857
- Mariotte, E., et al. (2016). Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency. The Lancet Haematology, 3(5), e237–e245.
- Zheng, X. L., Al-Housni, Z., Cataland, S. R., Coppo, P., Geldziler, B., Germini, F., Iorio, A., Keepanasseril, A., Masias, C., Matsumoto, M., McCrae, K. R., McIntyre, J., Mustafa, R. A., Peyvandi, F., Russell, L., Tarawneh, R., Vesely, S. K., & International Society on Thrombosis and Haemostasis (2025). 2025 focused update of the 2020 ISTH guidelines for management of thrombotic thrombocytopenic purpura. Journal of thrombosis and haemostasis : JTH, 23(11), 3711–3732. https://doi.org/10.1016/j.jtha.2025.06.002