Key Takeaways
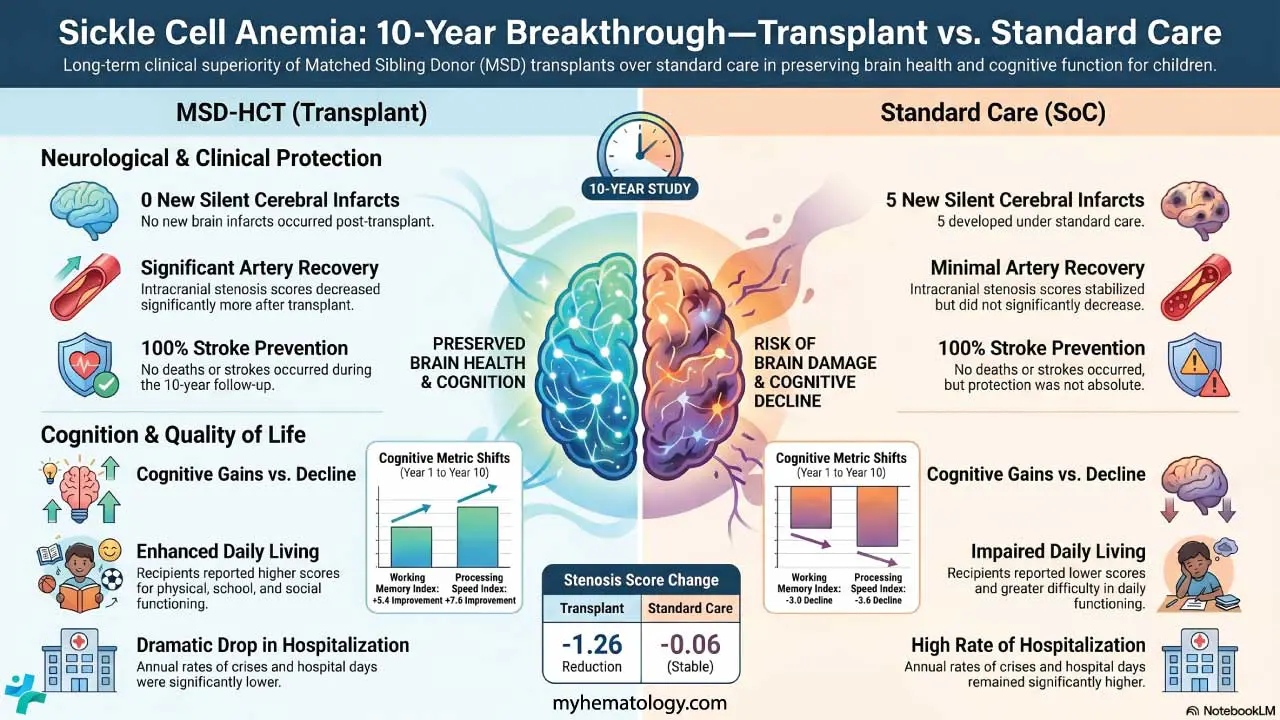
- Over 10 years, no new silent cerebral infarcts (painless patches of brain injury) developed in children who received a matched sibling transplant. In the standard-care group, 5 additional children developed new silent infarcts.
- Children in the transplant group scored an average of 96.5 on processing speed tests, compared to 83.7 in the standard-care group. Processing speed reflects how quickly the brain handles everyday information and learning tasks.
- Social quality of life scores were higher after transplantation (93.8 vs 86.4), suggesting that improved brain health may carry real benefits for how children interact with peers and engage in daily life.
- Both treatment pathways, when delivered in specialist centres with close monitoring, prevented major catastrophic events. The transplant advantage appeared most clearly in silent, cumulative brain protection over time.
The Problem No Parent Should Have to Face
Imagine being told that your child has a roughly one-in-ten chance of having a stroke before their 18th birthday. That is the reality for children living with sickle cell anaemia (SCA), a genetic blood disorder affecting millions of people worldwide.
In SCA, red blood cells that are normally soft and disc-shaped become rigid and crescent-like. These abnormal cells clump together, block small blood vessels, and deprive organs of oxygen. When this happens in the brain, it can cause a stroke. It can also cause something even more insidious: silent brain damage. No seizure. No paralysis. No obvious warning. Just small, painless areas of injury quietly forming inside a growing brain, slowly chipping away at memory, concentration, and learning ability.
For decades, the standard treatment for children at highest stroke risk has been regular blood transfusions — a management strategy that helps but comes with real costs. The body can develop antibodies against donated blood (called alloimmunisation), iron builds up in vital organs over time, and the child remains tethered to a lifelong treatment that never actually fixes the underlying problem.
A different path has long existed: a bone marrow transplant from a matched sibling donor, formally called hematopoietic cell transplantation (HCT). It is the only treatment that can fundamentally replace the faulty blood-making system with a healthy one. But for many years, the evidence comparing it head-to-head against standard care in high-risk children was limited. The DREPAGREFFE trial was designed to change that.
How Doctors Identify Children at Risk
Before unpacking the trial, it helps to understand how doctors detect which children are in danger. A painless bedside scan called Transcranial Doppler (TCD) ultrasound measures how fast blood flows through the brain's major arteries. When blood moves too quickly at 200 centimetres per second or above, it signals that an artery is narrowed and under strain. That narrowing dramatically raises stroke risk.
Children flagged by TCD are placed on a programme of regular blood transfusions to slow that flow down and reduce the danger. This strategy has already brought the rate of first strokes in SCA children down sharply from historical figures. But it is a holding strategy, not a cure. The question the DREPAGREFFE team asked was direct: can a transplant do better?
The DREPAGREFFE Trial: What Was Done and How
DREPAGREFFE-1 was France's first prospective trial to directly compare haematopoietic cell transplantation against standard care in children with SCA who were already receiving transfusions for high-risk TCD readings. Between December 2010 and June 2013, 67 children aged 5 to 15 years were enrolled across multiple French centres.
The trial used an approach called genetic randomisation. Children were assessed for whether a matched sibling donor — a brother or sister without SCA whose immune markers closely matched the patient — was available. If a matched donor existed (n=32), the child received a transplant. If not (n=35), the child continued on standard care: transfusions for at least one year, potentially switching to the drug hydroxyurea if blood-flow velocities normalised. This was not a coin-flip randomisation, but the availability of a matched sibling was essentially outside anyone's control, making it a reasonable natural comparison. Statistical methods including propensity-score matching were used to account for differences between the two groups.
Seven of the 67 children had a prior history of overt stroke at the time of enrolment. Results at one and three years had already been published, showing that blood-flow velocities in the brain were significantly lower after transplantation, vessel narrowing (stenosis) improved more in the transplant group, and physical and school-related quality of life scores were better [3,4]. But the critical question remained: would those early advantages hold up over a decade? And would they translate into real protection for the brain?
What 10 Years of Follow-Up Revealed
The 10-year data, now published in the American Journal of Hematology, paint a compelling picture.
The most striking finding concerns silent cerebral infarcts (SCIs). At the time of enrolment, there were actually more SCIs in the transplant group than in the standard-care group (12 versus 6). Yet over 10 years, not a single new silent infarct developed in the transplant arm. In the standard-care group, five additional children developed new SCIs (p = .010). In some patients in the transplant group, previously visible infarcts either disappeared or shrank to under 3 mm.
The cognitive differences were equally striking. Processing speed as in how quickly the brain can take in and act on information, a skill that underpins academic performance and daily function was significantly higher in children who had received a transplant. Their average score was 96.5, compared to 83.7 in the standard-care group (p = .035). Social quality of life scores followed the same pattern: 93.8 in the transplant group versus 86.4 in the standard-care arm (p = .003).
One finding that deserves particular attention: throughout the entire 10-year follow-up period, no deaths and no overt strokes occurred in either group. This is an important safety context. Both treatment pathways, when applied to monitored, high-risk children in a specialist setting, kept the most catastrophic outcomes at bay. The transplant, however, appeared to provide substantially better protection for the brain over the long term.
Why This Matters for Patients and Families
For families navigating a diagnosis of sickle cell anaemia, these results carry genuine weight. The first question most parents ask is not about velocities or stenosis scores. It is: will my child be okay? Will they go to school, make friends, keep up, live fully?
The DREPAGREFFE data suggest that for children who have a matched sibling donor and who are already at high stroke risk, a transplant does not just manage the disease but it may actively protect the developing brain in ways that transfusions and hydroxyurea alone cannot fully match over time.
This matters especially because brain injury in SCA so often goes unnoticed. A child with multiple silent infarcts may not appear obviously unwell. They may simply struggle a little more to concentrate, take slightly longer to learn new material, or find certain tasks harder than their peers. By the time those patterns become apparent, years of quiet damage may have already occurred. Prevention is what makes the transplant advantage so significant here.
The Limitations: What This Study Cannot Tell Us
Honest science includes its own caveats, and this study has several worth understanding.
The sample size was relatively small with only 67 children across both arms. Findings from a study this size should be treated as important evidence, not definitive proof for every clinical scenario. Second, because assignment depended on donor availability rather than coin-flip randomisation, residual differences between groups cannot be entirely ruled out, even with statistical adjustments. Third, the transplant used a myeloablative conditioning regimen meaning the child's existing bone marrow was destroyed before the new cells were infused. This process carries a well-documented risk of infertility, a consequence families must weigh carefully. Follow-up duration and completeness also varied across participants.
Perhaps most importantly, these results apply to a specific population: children with SS or sickle β0-thalassemia genotypes, abnormally high TCD velocities, access to specialist French sickle cell centres, and a matched sibling donor. The majority of children with SCA worldwide do not have a matched sibling donor available. What these findings mean for unrelated donor transplants, or for the newer gene therapy approaches now entering clinical use, remains an open and active area of research.
Where the Science Goes From Here
The DREPAGREFFE-2 trial (NCT05053932) is extending this work further, continuing to track long-term cerebrovascular outcomes and cognitive performance in the original cohort. Meanwhile, the field of SCA treatment is moving quickly. Gene therapies that modify or replace the faulty hemoglobin gene have received regulatory approvals in recent years, offering potential cure even for patients without a matched sibling donor. How those emerging options will compare to matched sibling HCT over 10 or 20 years is a question future trials will need to answer.
What the DREPAGREFFE trial has already given us is something rare and valuable in medicine: long-term prospective data. Not a short-term surrogate marker. Not a result extrapolated from a laboratory model. A decade of follow-up in real children, showing real differences in brain health and cognitive function.
The Takeaway
Sickle cell anaemia is not just a disease of pain crises and anaemia. It is a disease that can quietly injure the brain over years, altering the trajectory of a child's life before anyone notices. For the high-risk children who were part of DREPAGREFFE-1, those who received a matched sibling transplant showed better brain protection, fewer silent infarcts, faster cognitive processing, and higher social quality of life a full decade later.
No new strokes. No deaths. And a brain that, by the evidence, fared meaningfully better.
This study is a reminder that durable biological correction — when safely achievable — can carry advantages that only become fully visible over time. Short follow-up periods in clinical research can miss exactly this kind of long-term divergence. The DREPAGREFFE team's commitment to a 10-year endpoint is precisely what makes this work so instructive.
The decision about whether to pursue a transplant remains deeply personal, involving risks, family circumstances, donor availability, and values that no study can resolve on a family's behalf. But the evidence now available should make that conversation more informed, more honest, and more grounded than ever before.
Source: F. Bernaudin, S. Verlhac, E. Ducros-Miralles, et al., “Better 10-Year Cerebrovascular Outcome After Transplant Than on Standard-Care in Sickle Cell Anemia: DREPAGREFFE Trial,” American Journal of Hematology (2026): 1–19, https://doi.org/10.1002/ajh.70336.
References
- Bernaudin, F., Verlhac, S., Peffault de Latour, R., Dalle, J. H., Brousse, V., Petras, E., Thuret, I., Paillard, C., Neven, B., Galambrun, C., Divialle-Doumdo, L., Pondarré, C., Guitton, C., Missud, F., Runel, C., Jubert, C., Elana, G., Ducros-Miralles, E., Drain, E., Taïeb, O., … DREPAGREFFE Trial Investigators (2019). Association of Matched Sibling Donor Hematopoietic Stem Cell Transplantation With Transcranial Doppler Velocities in Children With Sickle Cell Anemia. JAMA, 321(3), 266–276. https://doi.org/10.1001/jama.2018.20059
- Kanter, J., Liem, R. I., Bernaudin, F., Bolaños-Meade, J., Fitzhugh, C. D., Hankins, J. S., Murad, M. H., Panepinto, J. A., Rondelli, D., Shenoy, S., Wagner, J., Walters, M. C., Woolford, T., Meerpohl, J. J., & Tisdale, J. (2021). American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood advances, 5(18), 3668–3689. https://doi.org/10.1182/bloodadvances.2021004394C
- Verlhac, S., Gabor, F., Paillard, C., Petras, M., Genty, I., Elmaleh, M., Thuret, I., Kamdem, A., Missud, F., Neven, B., Dalle, J. H., Peffault de Latour, R., & Bernaudin, F. (2021). Improved stenosis outcome in stroke-free sickle cell anemia children after transplantation compared to chronic transfusion. British Journal of Haematology, 193(1), 188–193. https://doi.org/10.1111/bjh.17204
- Bernaudin, F., Arnaud, C., Kamdem, A., Youn, J., Vasile, M., Hau, I., Madhi, F., Malterre, A., Delestrain, C., Epaud, R., Jung, C., & Verlhac, S. (2026). Early detection and management of extracranial arteriopathy reduces the incidence of silent cerebral infarcts in sickle cell anemia: a long-term prospective cohort study. Haematologica, 111(1), 314–328. https://doi.org/10.3324/haematol.2025.287720