Key Takeaways
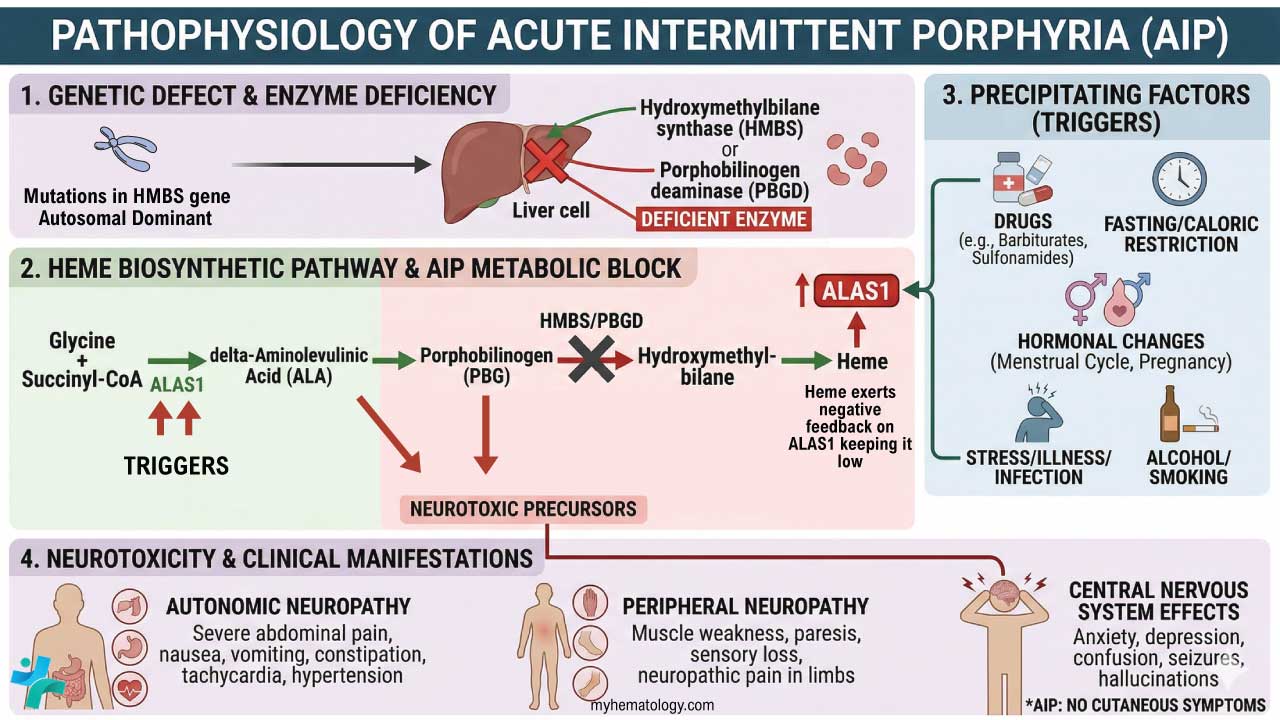
Hereditary spherocytosis is the most common inherited red blood cell membrane disorder, caused by faulty proteins that anchor the membrane skeleton. The most frequent cause is ankyrin deficiency, not spectrin deficiency as older sources suggest.
- Signs and symptoms ▾: Anemia, jaundice (yellow skin and eyes), and an enlarged spleen.
- Causes of hereditary spherocytosis ▾: Most common defect is a deficiency in ankyrin, a protein that anchors the membrane skeleton. This often causes a combined ankyrin–spectrin deficiency [1,4,11].
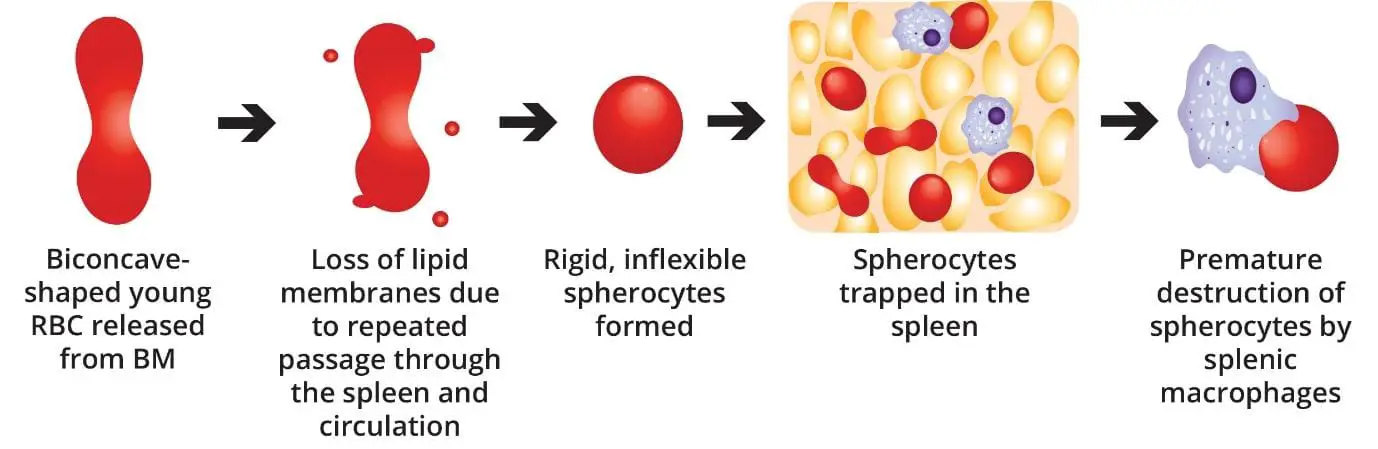
- Pathophysiology ▾: Normal-shaped young RBCs are produced by the bone marrow → repeated passage through the spleen and circulation leads to loss of membrane surface area releasing bilayer lipids in the form of cytoskeleton-free vesicles → spherocytes (rigid, inflexible and less deformable) → premature destruction of spherocytes by splenic macrophages (extravascular hemolysis).
- Laboratory Investigations ▾: Eosin-5'-maleimide (EMA) binding by flow cytometry, ideally paired with the cryohemolysis test [2,3].
- Treatment and management of hereditary spherocytosis ▾: Folic acid (a B vitamin needed to make red blood cells), transfusions when necessary, and surgical removal of the spleen in severe cases [1,5].
*Click ▾ for more information
What is Hereditary Spherocytosis?
Hereditary spherocytosis is an inherited disorder where red blood cells lose their normal disc shape. Healthy red blood cells look like flat discs with a dimple in the middle. In hereditary spherocytosis, they become small, round, and stiff. We call these misshapen cells spherocytes.
Spherocytes are fragile. The spleen filters them out of the bloodstream too early, before they have lived a full life of about 120 days. The early breakdown of red blood cells is called hemolysis. It leads to hemolytic anemia, which is anemia caused by red blood cells dying faster than the bone marrow can replace them [1,7].
Hereditary spherocytosis is the leading inherited cause of hemolysis in people of Northern European descent, but it is found in every population [1]. Severity varies a lot. Some people never have symptoms. Others have severe disease that shows up in the newborn period and needs regular transfusions [7,17].
Understanding Hemolytic Anemia
To understand hereditary spherocytosis, it helps to first understand hemolytic anemia. Hemolytic anemia is any anemia caused by red blood cells being destroyed too quickly. The destruction can happen in two places.
- Inside blood vessels (intravascular hemolysis): Red cells burst directly in the circulation. This happens with mechanical heart valves, severe malaria, or certain immune reactions.
- Outside blood vessels (extravascular hemolysis): Macrophages, the immune system's clean-up cells, engulf damaged red cells in the spleen and liver. This is the main mechanism in hereditary spherocytosis [1,7].
Comparison between Intravascular and Extravascular Hemolysis
| Feature | Intravascular Hemolysis | Extravascular Hemolysis |
| Where it happens | Inside blood vessels | Spleen, liver, and bone marrow |
| Mechanism | Direct rupture of red cells | Macrophages eat damaged red cells |
| Causes | Mechanical valves, severe malaria, paroxysmal nocturnal hemoglobinuria | Hereditary spherocytosis, sickle cell disease, warm autoimmune hemolytic anemia |
| Bilirubin pattern | Free hemoglobin in plasma; haptoglobin used up | Unconjugated bilirubin rises |
Inherited vs Acquired Hemolytic Anemia Causes
Hemolytic anemias fall into two broad groups. Inherited causes come from problems built into the red cell itself. These include membrane defects (such as hereditary spherocytosis), enzyme deficiencies (such as G6PD deficiency), and hemoglobin disorders (such as sickle cell disease and thalassemia).
Acquired causes come from outside the red cell. These include autoimmune attack, infections like malaria, mechanical damage, and drug reactions.
What Causes Hereditary Spherocytosis?
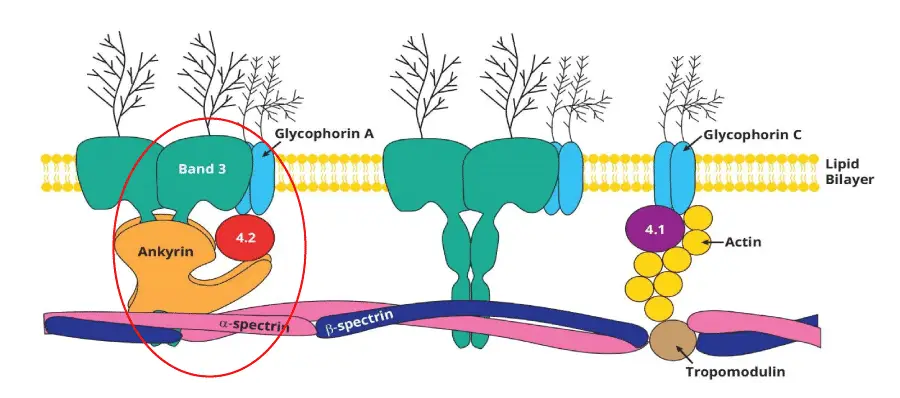
The cause of hereditary spherocytosis lies in the proteins that hold the red blood cell membrane together. Think of the membrane as a tent. The lipid bilayer is the canvas, and a network of proteins underneath acts like the poles and ropes. If those poles or ropes weaken, the canvas sags and pieces tear off.
Five genes account for almost all cases of hereditary spherocytosis [1,4,11]:
| Gene | Protein | How often it is involved | Inheritance |
| ANK1 | Ankyrin | About 50% | Dominant or recessive |
| SPTB | Beta-spectrin | About 20% | Dominant |
| SLC4A1 | Band 3 | About 15–20% | Dominant |
| SPTA1 | Alpha-spectrin | About 5% (severe form) | Recessive |
| EPB42 | Protein 4.2 | About 5% | Recessive |
About 75% of cases follow autosomal dominant inheritance. This means a child needs only one faulty copy of the gene from one parent to have the condition. The remaining 25% are either recessive (both parents pass on a faulty copy) or appear as brand new mutations with no family history [1,7]. The recessive forms, especially those involving SPTA1, tend to be more severe and may show up at birth [4].
Why the Gene Matters: Genotype–Phenotype Correlation
Recent research shows that the affected gene predicts how severe the disease will be [4,9]:
- Band 3 mutations (SLC4A1) usually cause mild to moderate disease.
- Ankyrin and beta-spectrin mutations are the most common and produce variable severity.
- Recessive alpha-spectrin mutations (SPTA1) cause severe, often transfusion- dependent disease. These patients are also more likely to need a second operation if they first have a partial splenectomy [9].
This is why genetic testing is becoming more common in unusual or severe cases. It helps doctors plan treatment.
Pathophysiology of Hereditary Spherocytosis
Red blood cells start out the right shape. The bone marrow produces them as smooth, flexible discs. The trouble in hereditary spherocytosis begins once they enter the circulation.
Every time a red cell squeezes through a tight space, the weakened membrane loses tiny pieces. These lost fragments are called microvesicles [1,7]. Over time, the cell loses surface area but keeps the same volume inside. It rounds up into a sphere.
The most punishing part of this journey is the spleen. To leave the splenic red pulp, red blood cells must squeeze through tiny gaps only 1 to 3 micrometers wide. The cells are also exposed to a harsh environment of low oxygen, low glucose, and low pH. This is sometimes called splenic conditioning [1,5].
The result is a vicious cycle:
- The cell loses membrane but not volume.
- It becomes a smaller, denser spherocyte.
- The spherocyte cannot bend enough to escape the spleen.
- Splenic macrophages destroy it.
This is why the spleen is the central villain in hereditary spherocytosis. It is also why removing the spleen is such an effective treatment.
How Severe Is Hereditary Spherocytosis?
Doctors classify hereditary spherocytosis by severity. The British Society for Haematology criteria (also known as the Bolton-Maggs criteria) are the most widely used [1,17]:
| Severity | Hemoglobin (g/dL) | Reticulocytes (%) | Bilirubin (μmol/L) | Transfusion need |
| Trait or very mild | Normal | < 3 | Normal | None |
| Mild | 11 - 15 | 3 - 6 | 17 - 34 | None or rare |
| Moderate | 8 - 11 | ≥ 6 | > 34 | Occasional |
| Severe | < 8 | > 10 | > 51 | Regular |
About 20–30% of patients have mild disease. Around 60–70% have moderate disease. Only 3–5% have severe disease [17]. This grading guides everything from whether to give folic acid to whether to remove the spleen.
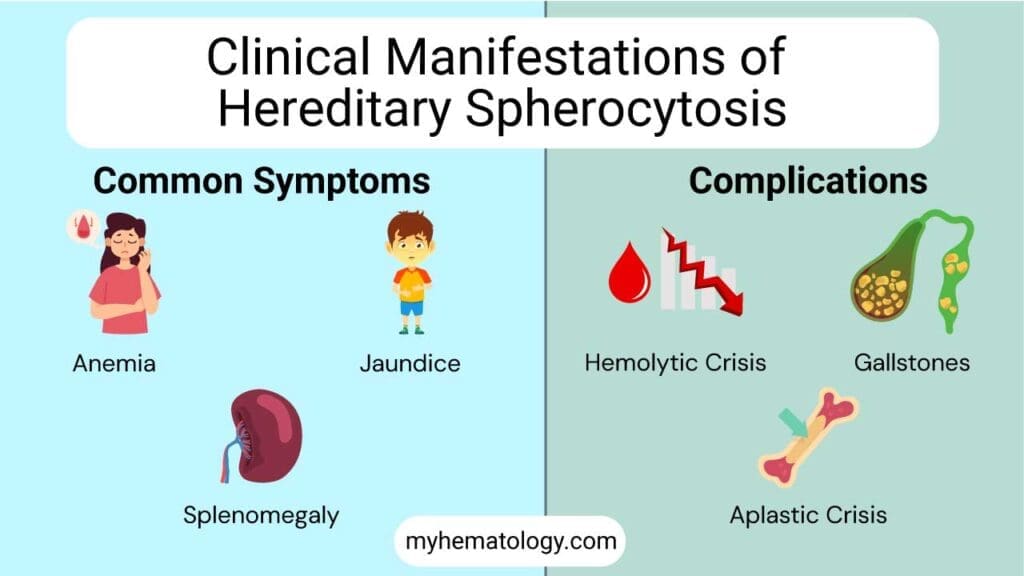
Signs and Symptoms of Hereditary Spherocytosis
Hereditary spherocytosis usually shows up as a classic triad of three findings.
- Anemia causes fatigue, pale skin, and shortness of breath on exertion. The body simply does not have enough red blood cells to carry oxygen efficiently.
- Jaundice is the yellow tinge in the skin and the whites of the eyes. It comes from a buildup of bilirubin, the yellow pigment released when red blood cells break down. Jaundice in hereditary spherocytosis often comes and goes, getting worse during infections, fatigue, or pregnancy.
- Splenomegaly is an enlarged spleen. Most patients have only mild to moderate enlargement. The size of the spleen alone is not a reason to operate [1].
Presentation in Newborns
Hereditary spherocytosis is an under-recognized cause of jaundice in newborns. Affected babies may need phototherapy (special blue light) within the first 24 hours of life. Some need an exchange transfusion, where the baby's blood is gradually replaced with donor blood [6,12]. Many also need transfusions in their first year. Erythropoietin, the hormone that tells the marrow to make red blood cells, can sometimes reduce or remove the need for transfusions in selected infants [6].
Crises in Hereditary Spherocytosis
Several types of crisis can affect patients with hereditary spherocytosis. Each is worth knowing.
- Hemolytic crisis: A sudden surge of red cell destruction, often triggered by an infection. Hemoglobin drops, jaundice deepens, and the spleen swells.
- Aplastic crisis: Almost always caused by parvovirus B19. This virus infects the cells in the bone marrow that make red blood cells and shuts down production for 7 to 10 days. Healthy people barely notice. But patients with hereditary spherocytosis depend on constant high red cell output, so their hemoglobin can plummet. Many need urgent transfusion [5,7].
- Megaloblastic crisis: Caused by running out of folate. Daily folic acid supplements prevent it [1].
Pigment Gallstones
Constantly breaking down red blood cells produces a lot of bilirubin. Bilirubin can crystallize in the gallbladder as pigment gallstones. These can develop even in childhood. When stones cause pain or infection, surgeons remove the gallbladder. They often do this at the same time as splenectomy [1,5].
Other Long-Term Issues
Severe untreated disease can cause growth delay, late puberty, and skeletal changes from an over-stretched bone marrow. Iron overload can also build up, even without regular transfusions, especially in patients who also carry mutations in the HFE iron-regulation gene. Doctors monitor ferritin, a blood marker of iron stores [1].
How is Hereditary Spherocytosis Diagnosed?
Diagnosing hereditary spherocytosis is a step-by-step process. It moves from confirming hemolysis, to ruling out immune causes, to running tests specific for the condition [2,3,8].
Step 1: Confirm Hemolysis
The first round of tests looks for evidence that red blood cells are being destroyed too fast.
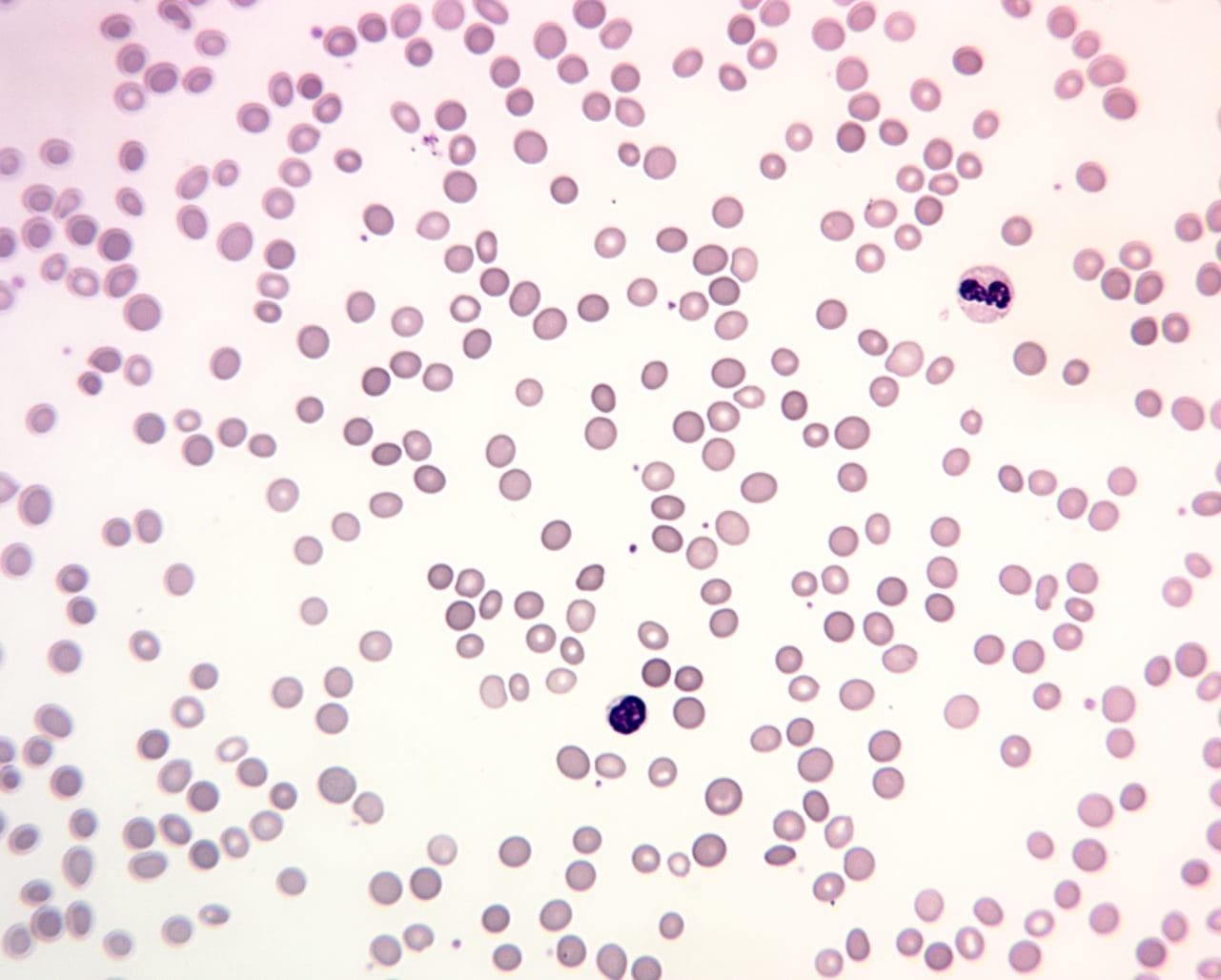
- Full blood count shows anemia and a high reticulocyte count (young red cells released from the marrow). The MCHC (mean corpuscular hemoglobin concentration) is often raised, because spherocytes are denser than normal cells. This is a strong early clue.
- Peripheral blood smear reveals spherocytes. They look small and dark, with no central pale spot.
- Biochemistry shows raised unconjugated bilirubin, raised LDH (an enzyme released from broken cells), and low haptoglobin (a protein that mops up free hemoglobin).
Step 2: Rule Out Immune Hemolysis
The direct antiglobulin test (DAT), also called the Coombs test, looks for antibodies stuck to red blood cells. In hereditary spherocytosis, the DAT must be negative. A positive DAT points instead to autoimmune hemolytic anemia, which can also produce spherocytes.
Step 3: Confirm Hereditary Spherocytosis
Once doctors have ruled out an immune cause, they run tests specific for hereditary spherocytosis.
| Test | What it measures | Status in 2024–2025 |
| EMA binding (flow cytometry) | How much fluorescent dye binds to band 3 protein | First-line confirmatory test [2,3] |
| Cryohemolysis test | Whether red cells lyse in cold conditions | Recommended alongside EMA for best sensitivity [2] |
| Osmotic fragility test | Whether red cells lyse in dilute saline | No longer first-line; misses about 25% of mild cases [2,3] |
| Acidified glycerol lysis test | Time to lyse in glycerol solution | Used where EMA is unavailable |
| Ektacytometry | Red cell flexibility under stress | Specialist test for unusual cases [8] |
Step 4: Genetic Testing in Selected Cases
Genetic testing is not routine. Doctors order it when [4,9]:
- The clinical picture is unusual.
- There is no family history.
- Recessive inheritance is suspected.
- The team is planning a partial splenectomy in a child with severe disease.
Differential Diagnosis
Several conditions can mimic hereditary spherocytosis. Doctors keep these in mind:
- Autoimmune hemolytic anemia (DAT positive)
- Hereditary stomatocytosis. Splenectomy is dangerous in this condition because of severe blood clot risk [1]
- Congenital dyserythropoietic anemia type II
- G6PD deficiency and unstable hemoglobins
- ABO hemolytic disease of the newborn in jaundiced babies can be valuable for confirming the diagnosis, especially in cases with an unclear family history or when other tests are equivocal.
Treatment and Management
There is no cure for the underlying gene defect. The goal of treatment is to control hemolysis, prevent crises, and avoid complications. The plan depends on how severe the disease is [1,5].
Supportive Care
Most patients do well with simple measures:
- Folic acid supplements (usually 1–5 mg per day) for moderate to severe disease, to prevent megaloblastic crisis. Mild cases may not need it [1].
- Regular monitoring of hemoglobin, reticulocytes, bilirubin, and ferritin.
- Abdominal ultrasound checks for gallstones.
- Vaccinations, including the annual flu shot and routine immunizations.
- Transfusions during severe anemia or a crisis.
Care for Newborns
Newborns with hereditary spherocytosis often need phototherapy for jaundice, following the 2022 American Academy of Pediatrics guidelines [12]. If bilirubin stays dangerously high, an exchange transfusion may be needed. Erythropoietin can reduce transfusion needs in babies under one year old [6].
Splenectomy: When and Why
Splenectomy is the surgical removal of the spleen. It is the most effective treatment for the chronic hemolysis of hereditary spherocytosis. By removing the main site where spherocytes are destroyed, splenectomy raises hemoglobin, eases symptoms, and improves quality of life [1,5,10].
It is indicated for:
- Severe disease (hemoglobin under 8 g/dL or transfusion dependence)
- Moderate disease with significant symptoms, growth failure, or other complications
- Symptomatic gallstones requiring cholecystectomy (often combined)
It is not routine for mild disease.
Timing matters. Splenectomy is usually delayed until after age 6 because young children face the highest risk of overwhelming infection without a spleen. Earlier surgery is sometimes necessary in severe transfusion-dependent disease.
Vaccinate before surgery, ideally at least 2 weeks ahead [14]:
- Pneumococcal vaccines (PCV13 or PCV15, then PPSV23)
- Meningococcal ACWY and Meningococcal B
- Haemophilus influenzae type b (Hib)
After surgery, patients need:
- Lifelong prophylactic penicillin V (or an alternative), at least until adulthood and often for life, especially in children [14]
- A medical alert bracelet
- Standby antibiotics for fever
- Clear education about the risks of infection
Partial vs. Total Splenectomy
Surgeons can remove the entire spleen (total splenectomy) or just part of it (partial splenectomy). Partial splenectomy keeps some splenic tissue alive to maintain immune function. In children, partial splenectomy raises hemoglobin by about 3 g/dL on average [9,10].
There is a trade-off. Between 10% and 25% of patients need a second operation later because the remaining splenic tissue grows back and hemolysis returns. Patients with SPTA1 mutations are especially likely to need this completion splenectomy, about 70% of the time [9]. This is one reason doctors increasingly use genetic testing to guide surgical planning.
Long-Term Risks of Splenectomy
The most feared complication after splenectomy is overwhelming post-splenectomy infection (OPSI), a rapid and severe bacterial infection. But there are other long-term risks too [13]:
- Blood clots, including in the deep veins and the portal vein of the liver
- Pulmonary hypertension (high blood pressure in the lungs)
- A small increase in heart and vascular disease
These risks are why splenectomy is reserved for patients who really need it.
Managing Specific Complications
- Gallstones: Symptomatic stones lead to cholecystectomy, usually combined with splenectomy if both are planned.
- Iron overload: Doctors monitor ferritin and may use iron chelation drugs, such as deferasirox, if iron builds up.
- Pregnancy: Hemoglobin tends to drop in pregnancy. Doctors monitor closely and continue folic acid. Most pregnancies go smoothly.
Living with Hereditary Spherocytosis
For students preparing to work with patients, and for caregivers supporting a loved one, the practical day-to-day matters as much as the science.
- Do not stop folic acid without medical advice.
- Seek medical care quickly for a fever, especially after splenectomy.
- Travel safely. Keep travel vaccinations up to date and carry standby antibiotics and a medical alert.
- Sports are usually fine. Contact sports may be cautioned in patients with a very enlarged spleen because of the small risk of splenic injury.
- Family screening helps. First-degree relatives benefit from a full blood count, reticulocyte count, and blood film, because hereditary spherocytosis often runs in families.
Most people with hereditary spherocytosis live full, active lives. With modern diagnosis, supportive care, and selective splenectomy, life expectancy is close to normal in mild and moderate disease [5,7]. Severe disease used to carry a heavier burden, but outcomes have improved a lot thanks to partial splenectomy, erythropoietin in infancy, and structured
infection prevention [5,9].
Frequently Asked Questions (FAQs)
What is the most common defect in hereditary spherocytosis?
The most common defect in hereditary spherocytosis is a deficiency in the spectrin protein. Spectrin is a crucial component of the red blood cell membrane, providing structural support and maintaining its normal biconcave shape. When spectrin is deficient or abnormal, the red blood cells become sphere-shaped, which makes them more susceptible to destruction by the spleen.
What four main complications can occur in patients with hereditary spherocytosis?
- Hemolytic anemia: This is the most common complication, resulting from the premature destruction of red blood cells by the spleen. Symptoms include fatigue, weakness, shortness of breath, and pallor.
- Splenomegaly: The spleen becomes enlarged due to its increased workload in destroying damaged red blood cells. This can cause abdominal discomfort, fullness, and sometimes pain.
- Gallstones (Cholelithiasis): The breakdown of red blood cells releases bilirubin, which can accumulate in the gallbladder and form stones. This can lead to abdominal pain, nausea, and vomiting.
- Aplastic crisis: In rare cases, patients with hereditary spherocytosis may experience a sudden decrease in red blood cell production, resulting in a severe form of anemia. This can require blood transfusions or other interventions.
Is hereditary spherocytosis the same in everyone?
No. About 20–30% of patients have mild disease, 60–70% have moderate disease, and 3–5% have severe disease [17]. Severity depends on which membrane protein is affected and whether one or both copies of the gene carry a mutation [4]. If one parent has hereditary spherocytosis, what is the chance my child will inherit it? About 50% if the parent has the dominant form, which covers around 75% of all cases. The rest are recessive (both parents must carry a mutation) or appear as new mutations with no family history [1,7]. Genetic counseling helps families understand their risk.
Why remove the spleen ifit is just doing its normal job?
The spleen normally clears damaged red cells. In hereditary spherocytosis, it destroys cells that are weak but still useful. Removing the spleen lets those cells survive long enough to do their job. The genetic defect remains, so the cells stay spherical, but they last much longer [5].
What infections do people worry about after splenectomy?
The main worry is encapsulated bacteria: Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b. Vaccines, daily preventive antibiotics (especially in children), and standby antibiotics for fever all reduce this risk [14].
Why do people with hereditary spherocytosis get gallstones?
Constant red cell breakdown produces extra bilirubin, which can crystallize in the gallbladder as pigment gallstones. They may appear in childhood. Surgery is needed only if the stones cause symptoms [1,5].
Can someone with hereditary spherocytosis live a normal life?
Yes. With folic acid (in moderate to severe disease), regular monitoring, and selective splenectomy, most patients
live full and active lives. Pregnancy, school, work, and sports are usually unaffected [5,7].
Glossary of Related Medical Terms
- Anemia — A condition in which the number of red blood cells, or the amount of hemoglobin in them, is below normal, reducing the blood's ability to carry oxygen.
- Ankyrin (ANK1) — A protein that anchors the red blood cell's internal skeleton to its outer membrane. Mutations in the ANK1 gene are the most common cause of hereditary spherocytosis.
- Aplastic crisis — A temporary shutdown of red blood cell production by the bone marrow, usually triggered by parvovirus B19 infection, causing a sudden severe drop in hemoglobin.
- Autosomal dominant inheritance — A genetic pattern in which a single copy of an abnormal gene from one parent is enough to cause the condition.
- Band 3 protein (SLC4A1) — A red blood cell membrane protein that exchanges chloride and bicarbonate ions and helps anchor the cell skeleton; the target of the EMA dye used in diagnosis.
- Bilirubin — A yellow pigment produced when hemoglobin from broken-down red blood cells is processed. High levels cause jaundice.
- Cholecystectomy — Surgical removal of the gallbladder, often performed if pigment gallstones cause symptoms.
- Cryohemolysis test — A laboratory test in which red blood cells are exposed to cold conditions; spherocytes break down more readily and the test is used alongside EMA binding to confirm HS.
- Direct Antiglobulin Test (DAT) / Coombs test — A blood test that detects antibodies attached to red blood cells. A negative DAT helps distinguish HS from autoimmune hemolytic anemia.
- Ektacytometry — A specialized test that measures how easily red blood cells deform under stress; produces a characteristic curve in HS.
- EMA (eosin-5'-maleimide) binding test — A flow cytometry test that uses a fluorescent dye binding to band 3 protein. Reduced fluorescence indicates HS; this is now the preferred confirmatory test.
- Extravascular hemolysis — Destruction of red blood cells outside the blood vessels, mostly in the spleen and liver. The dominant mechanism in HS.
- Folic acid (folate) — A B-vitamin essential for red blood cell production; supplemented in HS to keep up with high red-cell turnover.
- Hemoglobin — The iron-containing protein in red blood cells that carries oxygen.
- Hemolysis — The breakdown of red blood cells.
- Hemolytic crisis — A sudden episode of accelerated red blood cell destruction, usually triggered by infection, leading to worsening anemia and jaundice.
- Jaundice — Yellowing of the skin and the whites of the eyes due to elevated bilirubin.
- Mean corpuscular hemoglobin concentration (MCHC) — A red blood cell index. Elevated MCHC is a classic clue for HS because spherocytes are densely packed with hemoglobin.
- Osmotic fragility test — An older test in which red cells are placed in dilute salt solutions; spherocytes burst more easily. Now considered less sensitive than EMA binding.
- Overwhelming post-splenectomy infection (OPSI) — A rare but life-threatening bacterial infection that can occur after spleen removal, typically caused by encapsulated bacteria.
- Parvovirus B19 — A virus that infects red-cell precursors in the bone marrow and can trigger an aplastic crisis in HS.
- Pigment gallstones — Gallstones formed from excess bilirubin; common in chronic hemolysis.
- Polychromasia — A bluish tinge of red blood cells on a smear, indicating young, recently released cells (reticulocytes).
- Reticulocyte — An immature red blood cell. A high reticulocyte count signals the marrow's effort to replace destroyed red cells.
- Spectrin (SPTA1, SPTB) — Long, flexible proteins that form the main scaffolding of the red cell membrane.
- Spherocyte — A small, round, dense red blood cell that has lost its central pale area; the diagnostic hallmark of HS on a smear.
- Splenectomy — Surgical removal of the spleen. May be partial (subtotal) or total.
- Splenomegaly — Enlargement of the spleen.
- Unconjugated bilirubin — The form of bilirubin produced from hemoglobin breakdown before the liver processes it; raised in extravascular hemolysis.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Bolton-Maggs, P. H., Langer, J. C., Iolascon, A., Tittensor, P., King, M. J., & General Haematology Task Force of the British Committee for Standards in Haematology (2012). Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. British journal of haematology, 156(1), 37–49. https://doi.org/10.1111/j.1365-2141.2011.08921.x
- Polizzi, A., Dicembre, L. P., Failla, C., Matola, T. D., Moretti, M., Ranieri, S. C., Papa, F., Cenci, A. M., & Buttarello, M. (2025). Overview on Hereditary Spherocytosis Diagnosis. International journal of laboratory hematology, 47(1), 18–25. https://doi.org/10.1111/ijlh.14376
- Wu, Y., Liao, L., & Lin, F. (2021). The diagnostic protocol for hereditary spherocytosis-2021 update. Journal of clinical laboratory analysis, 35(12), e24034. https://doi.org/10.1002/jcla.24034
- Tole, S., Dhir, P., Pugi, J., Drury, L. J., Butchart, S., Fantauzzi, M., Langer, J. C., Baker, J. M., Blanchette, V. S., Kirby-Allen, M., & Carcao, M. D. (2020). Genotype-phenotype correlation in children with hereditary spherocytosis. British journal of haematology, 191(3), 486–496. https://doi.org/10.1111/bjh.16750
- Turpaev, K., Bovt, E., Shakhidzhanov, S., Sinauridze, E., Smetanina, N., Koleva, L., Kushnir, N., Suvorova, A., & Ataullakhanov, F. (2025). An overview of hereditary spherocytosis and the curative effects of splenectomy. Frontiers in physiology, 16, 1497588. https://doi.org/10.3389/fphys.2025.1497588
- Achenjang, N.-S., Jadczak, E., Ryan, R. M., & Nock, M. L. (2025). Hereditary Spherocytosis: Review of Presentation at Birth. Children, 12(9), 1207. https://doi.org/10.3390/children12091207
- Zamora EA, Schaefer CA. Hereditary Spherocytosis. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539797/
- Kalfa T. A. (2021). Diagnosis and clinical management of red cell membrane disorders. Hematology. American Society of Hematology. Education Program, 2021(1), 331–340. https://doi.org/10.1182/hematology.2021000265
- Ramjist, J. K., Dubljevic, T., Lapidus-Krol, E., Grace, R. F., Heeney, M. M., Oni, M. O., Towerman, A., Davidoff, A., Takemoto, C., Brown, R. L., Brungardt, J., Beaman, M., Rice, H. E., Nichols, S., Saadai, P., Carcao, M., & Langer, J. C. (2025). Correlation of Genetic Mutation With Outcomes in Children With Hereditary Spherocytosis Undergoing Partial Splenectomy: A Multicentre Study. Journal of pediatric surgery, 60(4), 162229. https://doi.org/10.1016/j.jpedsurg.2025.162229
- Rice, H. E., Englum, B. R., Rothman, J., Leonard, S., Reiter, A., Thornburg, C., Brindle, M., Wright, N., Heeney, M. M., Smithers, C., Brown, R. L., Kalfa, T., Langer, J. C., Cada, M., Oldham, K. T., Scott, J. P., St Peter, S., Sharma, M., Davidoff, A. M., Nottage, K., … Splenectomy in Congenital Hemolytic Anemia (SICHA) Consortium (2015). Clinical outcomes of splenectomy in children: report of the splenectomy in congenital hemolytic anemia registry. American journal of hematology, 90(3), 187–192. https://doi.org/10.1002/ajh.23888
- Iolascon, A., Andolfo, I., & Russo, R. (2019). Advances in understanding the pathogenesis of red cell membrane disorders. British journal of haematology, 187(1), 13–24. https://doi.org/10.1111/bjh.16126
- Kemper, A. R., Newman, T. B., Slaughter, J. L., Maisels, M. J., Watchko, J. F., Downs, S. M., Grout, R. W., Bundy, D. G., Stark, A. R., Bogen, D. L., Holmes, A. V., Feldman-Winter, L. B., Bhutani, V. K., Brown, S. R., Maradiaga Panayotti, G. M., Okechukwu, K., Rappo, P. D., & Russell, T. L. (2022). Clinical Practice Guideline Revision: Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics, 150(3), e2022058859. https://doi.org/10.1542/peds.2022-058859
- Schilling, R. F., Gangnon, R. E., & Traver, M. I. (2008). Delayed adverse vascular events after splenectomy in hereditary spherocytosis. Journal of thrombosis and haemostasis : JTH, 6(8), 1289–1295. https://doi.org/10.1111/j.1538-7836.2008.03024.x
- Davies, J. M., Lewis, M. P., Wimperis, J., Rafi, I., Ladhani, S., Bolton-Maggs, P. H., & British Committee for Standards in Haematology (2011). Review of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen: prepared on behalf of the British Committee for Standards in Haematology by a working party of the Haemato-Oncology task force. British journal of haematology, 155(3), 308–317. https://doi.org/10.1111/j.1365-2141.2011.08843.x
- Management of Anemia: A Comprehensive Guide for Clinicians (Provenzano et al., 2018)
- Goldberg S, Hoffman J. Clinical Hematology Made Ridiculously Simple, 1st Edition: An Incredibly Easy Way to Learn for Medical, Nursing, PA Students, and General Practitioners (MedMaster Medical Books). 2021.
- National Library of Medicine. (n.d.). Hereditary spherocytosis. MedlinePlus Genetics.