Introduction
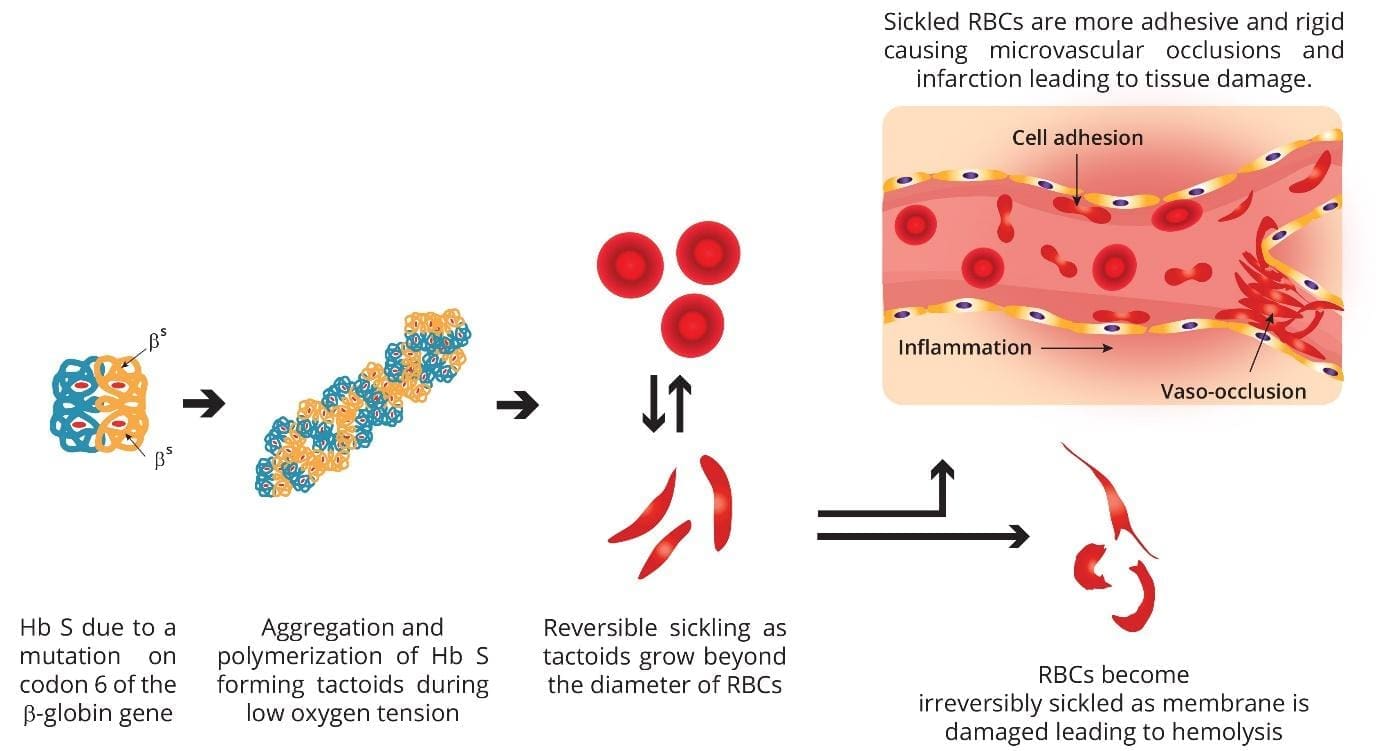
SCD arises from a single nucleotide mutation in the beta-globin gene (HBB) on chromosome 11. This mutation leads to the substitution of valine for glutamic acid at the sixth amino acid position of the beta-globin chain in the hemoglobin molecule. This mutation results in the abnormal hemoglobin S (HbS). The mutated β-globin chain forms abnormal hemoglobins called HbS. HbS molecules have a tendency to polymerize under low oxygen conditions, leading to the characteristic sickle shape of the red blood cells.
SCD is the most common inherited blood disorder globally, affecting millions of people, especially those of African descent. The prevalence varies geographically, with the highest rates in sub-Saharan Africa, followed by the Caribbean, India, and parts of the Americas. In the United States, approximately 100,000 individuals have SCD, and around 1 in 400 African American newborns have the disease.
SCD follows an autosomal recessive inheritance pattern. This means that individuals must inherit two copies of the HbS gene (one from each parent) to develop the disease. If only one parent carries the HbS gene, the individual will be a carrier (HbAS), exhibiting no symptoms but able to pass the gene to their offspring.
HbS molecules tend to polymerize and form rigid, insoluble crystals within red blood cells when they lose oxygen. This process causes the cells to deform into the characteristic sickle shape. Sickled red blood cells are less flexible and prone to getting stuck in small blood vessels, leading to blockages and reduced blood flow. This impaired blood flow and oxygen delivery contribute to the various symptoms and complications of SCD.
The symptoms of SCD vary depending on the severity and genotype. Some common symptoms include fatigue, weakness, shortness of breath, pale skin. Vaso-occlusive crises can cause episodes of severe pain, typically in the bones, joints, abdomen, or chest. Delayed growth and development in children can occur. Other symptoms include frequent infections, vision problems, stroke and pulmonary complications, like acute chest syndrome.
Diagnosis of SCD involves a combination of clinical features and laboratory tests, including complete blood count (CBC) which can reveal anemia and abnormal red blood cell morphology. Specific tests like hemoglobin electrophoresis identify the presence of HbS and other abnormal hemoglobin variants. While genetic testing confirms the presence of the HbS gene mutation. Other tests may be needed to assess complications like infections or organ damage.
While there is no cure for SCD, several treatment options can help manage the symptoms and prevent complications. Hydroxyurea (HU) is a medication that increases fetal hemoglobin levels (HbF) and reduces red blood cell production, leading to fewer painful episodes and improved quality of life. Blood transfusions can be used to treat severe anemia or complications like acute chest syndrome. Medications and non-pharmacological approaches like heat therapy and massage can help manage pain episodes. SCD patients are at increased risk of infections, so keeping vaccinations up-to-date is crucial. A potentially curative option for some young patients with severe SCD is bone marrow transplantation but it is high-risk as the need to get a HLA-compatible donor as well as there is a risk of graft versus host rejection. Last but not least, nutrition counseling, mental health support, and social support are essential for managing the chronic and complex nature of SCD.
Research into new treatment options for SCD is ongoing, including gene therapy and stem cell therapy. Additionally, efforts are focused on improving access to care and education for patients with SCD.
Hydroxyurea (HU) is a medication originally used as an anti-cancer agent. In the context of sickle cell disease (SCD), it acts as a disease-modifying therapy, meaning it can alter the course of the disease and improve long-term outcomes.
The journey of HU in SCD treatment began in the late 1960s. Researchers observed that HU could induce fetal hemoglobin (HbF) production in adults. HbF, present in babies but typically disappearing after birth, has a different structure than adult hemoglobin (HbA) and is less prone to sickling. This sparked the idea of using HU to increase HbF levels in SCD patients, potentially reducing sickling and its associated complications.
Prior to HU, SCD management primarily relied on supportive measures like pain management, blood transfusions, and antibiotics to address complications. HU introduced a paradigm shift by actively targeting the underlying disease mechanism. HU significantly decreases the frequency and severity of painful episodes, a hallmark of SCD. This is primarily due to the increase in HbF, which makes red blood cells more flexible and less prone to sickling. By reducing pain crises and other complications, HU improves patients' overall quality of life. They experience less fatigue, fewer hospitalizations, and increased ability to participate in daily activities. HU can help prevent or delay serious complications like acute chest syndrome, stroke, and organ damage. This translates to improved long-term health outcomes and potentially even increased lifespan.
HU's beneficial effects in SCD are attributed to several mechanisms, for example, increased HbF production by inhibiting ribonucleotide reductase, an enzyme involved in DNA synthesis. This indirectly stimulates the production of HbF. HU suppresses bone marrow activity, leading to fewer red blood cells being produced. This minimizes the number of potentially sickling cells. And HU has anti-inflammatory properties, helping to reduce the chronic inflammation associated with SCD complications.
HU has become a cornerstone of SCD treatment, significantly improving the lives of millions of patients worldwide. It is considered the only widely available medication with disease-modifying properties in SCD. While it's not a cure, it offers a powerful tool to manage the disease, reduce complications, and enhance quality of life. However, challenges remain. Underutilization of HU due to provider and patient concerns, side effects, and access limitations necessitate ongoing efforts to improve awareness, education, and accessibility of this revolutionary therapy.
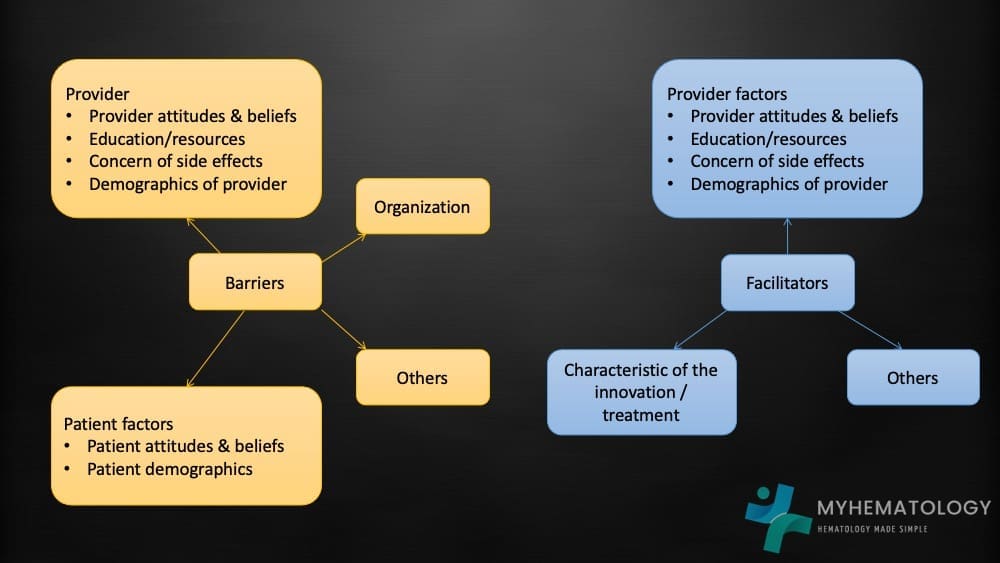
Recently Pizzo et al.'s paper published in the British Journal of Haematology explored the concerns of underutilization of hydroxyurea (HU) in sickle cell disease (SCD) management despite concrete evidence of the effectiveness of HU in amelioration of the disease, analyzing both barriers and facilitators that influence its prescription and patient adherence. The authors highlighted that the barriers to HU utilization include provider-related issues for example limited knowledge and awareness. Some healthcare providers lack sufficient knowledge about HU's benefits and effectiveness, leading to under-prescribing or hesitant initial prescriptions. Some providers have concerns about side effects like carcinogens as well as sterility issues leading to cautious or delayed initiation. Busy schedules and inadequate time for patient consultation can also hinder thorough explanations and monitoring of HU therapy.
Patient-related barriers include misinformation and mistrust as concerns and misconceptions about HU, often fueled by misinformation circulating in communities, can lead to patient reluctance or non-adherence. Fear of side effects similar to providers, may cause patients to harbor anxieties about potential side effects, deterring them from starting or continuing HU therapy. Poor understanding of SCD and HU's role in management can lead to decreased motivation and adherence to medication regimes.
To increase the uptake of HU among sickle cell patients, the critical role of comprehensive and engaging educational materials in optimizing HU adherence and combating misinformation in sickle cell disease (SCD) management cannot be ignored. While the potential of HU to revolutionize SCD management is undeniable, its underutilization persists. Thus, addressing this gap requires a powerful tool which is accessible, culturally relevant, and interactive educational resources tailored to both healthcare providers and patients.
These resources can empower providers by enhancing their knowledge and confidence in prescribing and managing HU through training and educational tools. They can also equip patients by combating misinformation and instill understanding about HU's benefits, potential side effects, and the importance of adherence. Finally, boosting motivation by utilizing engaging formats to capture attention, personalize information, and encourage proactive engagement with HU therapy.
By bridging the knowledge gap and fostering trust, comprehensive and engaging educational materials can unlock the full potential of HU, leading to improved adherence, enhanced quality of life, and ultimately, better health outcomes for individuals living with SCD.
Addressing Misinformation and Improving Adherence of HU
The fight against misinformation in SCD management, particularly regarding HU, plays a critical role in improving patient adherence and health outcomes. There are exaggerated claims about severe and common side effects of HU, like infertility, cancer, and organ damage, which can deter patients from starting or continuing therapy. The fear of debilitating side effects can lead to treatment abandonment, jeopardizing the potential benefits of HU and potentially worsening disease progression. Confusion surrounding how HU works and its long-term effects can fuel distrust and anxieties about unknown consequences, which can lead to non-adherence as patients may not see the direct connection between taking the medication and managing their symptoms.
The need for accurate and accessible information resources becomes paramount in this scenario. We need to develop culturally-sensitive educational materials and tailor information to address specific cultural beliefs and misconceptions about SCD and HU. Employ diverse channels like infographics, videos, and interactive apps to cater to different learning styles and preferences, even age and gender. We need to equip healthcare professionals with communication skills and resources to effectively address patient concerns and counter misinformation. Health practitioners should promote trusted sources of information by encouraging patients to rely on credible sources like medical organizations and healthcare professionals for accurate information rather than just the internet. Healthcare providers should also facilitate open communication to foster an environment where patients feel comfortable asking questions and expressing concerns about HU, allowing for transparent and trust-building dialogue.
By actively addressing misinformation and providing accessible, accurate information, we can empower patients to make informed decisions about HU, ultimately leading to improved adherence and better health outcomes for individuals living with SCD.
Patient education lies at the heart of optimizing hydroxyurea (HU) adherence in sickle cell disease (SCD) management. Understanding the benefits of HU, along with its potential side effects, can empower patients to actively participate in their own care and make informed decisions about their medication. It is important to constantly highlight the benefits of HU adherence to the patients. Studies consistently show that HU significantly decreases the frequency and severity of painful episodes, a major burden for SCD patients. This translates to improved quality of life, increased productivity, and reduced emotional distress. By preventing complications like acute chest syndrome and strokes, HU adherence leads to fewer hospital admissions and emergency room visits. This reduces healthcare costs and improves overall well-being. Beyond pain reduction, HU adherence can improve sleep quality, energy levels, and overall physical and mental health. Patients can engage in activities they may have previously avoided due to fatigue or fear of pain. Some research suggests that consistent HU use may even improve long-term survival for SCD patients, highlighting the crucial role of adherence in optimizing life expectancy.
Recommendation for Engaging Educational Materials
In the fight to improve HU adherence in SCD, traditional text-heavy materials often fall short, especially when trying to reach younger audiences and individuals with diverse learning styles. We need to offer more innovative formats like comics, infographics, and short videos, offering a more engaging and effective approach to patient education.
Visual storytelling transcends language barriers and literacy levels, making information accessible to everyone. Complex concepts can be broken down into simple narratives and visually compelling scenes like in comics, aiding comprehension and retention. Engaging characters and storylines leave a lasting impression, ensuring that key messages stick in the minds of patients.
Infographics is another innovative choice that is growing in popularity. Key information is condensed into visually appealing graphics and diagrams, preventing information overload and simplifying complex topics. Data and statistics are presented in a clear and easy-to-understand format in infographics, fostering comprehension and informed decision-making. The visual elements guide attention to crucial points, ensuring that patients grasp the most important aspects of HU therapy.
Although more difficult to curate, short videos can provide high engagement. Animated characters and real-life testimonials capture attention and create an emotional connection, fostering active learning and interest. Diverse video formats, from animations to interviews, cater to different preferences and even culture, and keep the learning process dynamic. Videos can be subtitled and adapted for various abilities, ensuring information reaches a wider audience.
These formats go beyond simply making information accessible. They actively engage patients, enhancing understanding and promoting long-term retention of crucial knowledge. The visual narratives and interactive elements of comics, infographics, and short videos provide a unique opportunity to address fears and concerns in sensitive topics like side effects that can be tackled in a gentle and empathetic way, fostering trust and reducing anxieties. Patient testimonials and success stories can empower individuals to take control of their health and embrace proactive HU adherence. Clear instructions and reminders about medication schedules and lifestyle modifications can be incorporated seamlessly into the storytelling, encouraging consistent adherence.
By embracing these engaging formats and tailoring them to different age groups and cultural contexts, we can revolutionize SCD education and empower patients to make informed decisions about HU. This, in turn, will pave the way for improved adherence, enhanced quality of life, and ultimately, better health outcomes for individuals living with SCD.
Empowering Adherence: A Mobile App for SCD Management
While engaging educational materials like comics and infographics can significantly improve HU adherence in SCD, the digital age offers another transformative tool: a comprehensive mobile app specifically designed for SCD patients. Such an app could address key adherence challenges by providing interactive modules on SCD basics, HU's mechanism of action, and its benefits (pain reduction, fewer hospitalizations, improved quality of life). Information on potential side effects presented transparently and encouragingly, addressing common concerns and misinformation. Culturally relevant content and personalized learning pathways can be provided to cater to diverse needs and preferences.
Another benefit of a mobile app for SCD is customizable alerts and notifications to ensure timely medication intake and avoid missed doses. Adherence tracking tools to visualize progress, celebrate successes, and identify areas for improvement which can be also viewed by healthcare providers and related family members. Gamification elements and rewards can be included to increase motivation and foster long-term adherence.
However, secure access to personal medical records, lab results, and treatment plans is necessary in the development of these apps. Features like symptom tracking tools to monitor pain levels, fatigue, and other symptoms, allowing for proactive adjustments in therapy. Personalized health insights and reports based on individual data can empower patients to take control of their health. Secure messaging features to directly communicate with healthcare professionals about questions, concerns, or unexpected symptoms is crucial. Appointment scheduling and telehealth consultations for increased convenience and improved access to care. While peer-to-peer support groups within the app connect users with other SCD patients for motivation and information exchange is also important .
This comprehensive mobile app can revolutionize SCD management by addressing knowledge gaps and combating misinformation. Patients can be equipped with accurate and accessible information, dismantling barriers to informed decision-making. The app can also promote proactive self-management by empowering patients to track their health, identify potential issues early, and adjust their behavior accordingly. Provider-patient communication can be strengthened by bridging the communication gap, enhancing trust, and facilitating timely interventions for optimal treatment outcomes. These will potentially reduce missed doses, improve treatment effectiveness, and ultimately lead to a healthier and more fulfilling life for individuals with SCD.
By leveraging the power of mobile technology, we can create a personalized and empowering tool that empowers patients, optimizes HU adherence, and transforms the landscape of SCD management. There are several SCD-related mobile apps in the market and these should be promoted to healthcare providers as well as patients to use.
Education as the Key to Hydroxyurea Adherence in SCD
The fight against sickle cell disease (SCD) hinges on optimal utilization of hydroxyurea (HU), a game-changing medication capable of revolutionizing quality of life. However, underutilization remains a stark reality, fueled by a potent cocktail of misinformation and inadequate patient education. Addressing these barriers is crucial to unlock the full potential of HU and improve health outcomes for millions living with SCD.
Traditional educational materials often fall short, failing to engage diverse audiences and effectively counter the misinformation swirling around HU. This is where innovative formats like comics, infographics, and mobile apps take center stage. Their captivating visuals, interactive elements, and personalized approach have the power to break down information into relatable narratives and visually compelling scenes, ensuring comprehension and retention; address anxieties and fears head-on, providing accurate and accessible information to dismantle harmful myths and rumors; foster self-efficacy and active participation in their own care by promoting informed decision-making and adherence behaviors; and create a platform for open dialogue between patients and healthcare providers, strengthening trust and enabling timely interventions.
The potential of these tools is immense. A mobile app equipped with educational modules, medication reminders, and personalized health updates can be a constant companion, empowering patients to manage their SCD proactively and optimize HU adherence.
The call to action is clear: researchers and healthcare professionals must invest in innovative educational approaches. By embracing comics, infographics, mobile apps, and other engaging formats, we can equip patients with the knowledge and confidence they need to embrace HU therapy, unleash its potential, and rewrite the narrative of SCD management. Let us prioritize patient education, dismantle misinformation, and empower individuals to conquer their disease, one informed decision at a time.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Pizzo A, Porter JS, Carroll Y, Burcheri A, Smeltzer MP, Beestrum M, Nwosu C, Badawy SM, Hankins JS, Klesges LM, Alberts NM. Provider prescription of hydroxyurea in youth and adults with sickle cell disease: A review of prescription barriers and facilitators. Br J Haematol. 2023 Dec;203(5):712-721. doi: 10.1111/bjh.19099. Epub 2023 Sep 10. PMID: 37691131.
- Cisneros GS, Thein SL. Recent Advances in the Treatment of Sickle Cell Disease. Front. Physiol., Sec. Red Blood Cell Physiology Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00435
- Williams TN, Thein SL. Sickle Cell Anemia and Its Phenotypes. Annu Rev Genomics Hum Genet. 2018 Aug 31;19:113-147. doi: 10.1146/annurev-genom-083117-021320. Epub 2018 Apr 11. PMID: 29641911; PMCID: PMC7613509.
- Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010; 3(3): 255–260.
- Badawy SM, Thompson AA, Lai JS, Penedo FJ, Rychlik K, Liem RI. Adherence to hydroxyurea, health-related quality of life domains, and patients' perceptions of sickle cell disease and hydroxyurea: a cross-sectional study in adolescents and young adults. Health Qual Life Outcomes. 2017; 15(1): 136.