Key Takeaways
Bernard-Soulier syndrome is inherited bleeding disorder characterized by large platelets, thrombocytopenia and impaired platelet function.
Causes ▾: Mutations in GP1BA, GP1BB, or GP9 genes resulting in defects in the glycoprotein Ib-IX-V complex on platelet surface
Pathophysiology ▾: Defective glycoprotein Ib-IX-V complex on platelet surface causes impaired platelet adhesion to von Willebrand factor. Thus a normal platelet plug is not able to be formed when an injury occurs.
Symptoms ▾: Increased bleeding tendency (variable severity) which typically presents in infancy or early childhood for example, epistaxis, gum bleeding and easy bruising.
Investigations ▾: Thrombocytopenia in CBC, macrothrombocytopenia in peripheral blood smear, prolonged bleeding time, reduced ristocetin-induced platelet aggregation.
Differential Diagnosis ▾: ITP, VWD, Glanzmann thrombasthenia
Treatment and Management ▾: Focuses on preventing/controlling bleeding including local hemostatic measures, platelet transfusions, recombinant activated factor VIIa (rFVIIa) and antifibrinolytics. Prognosis generally good with proper management
*Click ▾ for more information
Introduction
Bernard-Soulier syndrome (BSS) is a rare, inherited bleeding disorder characterized by:
- Unusually large platelets (macrothrombocytopenia)
- A low platelet count (thrombocytopenia)
- A defect in platelet function
This defect is specifically related to a deficiency or abnormality in a protein complex called glycoprotein Ib-IX-V (GPIb-IX-V) on the surface of platelets. This complex is crucial for platelets to stick to damaged blood vessels and form blood clots.
Causes of Bernard-Soulider Syndrome
Bernard-Soulier syndrome is caused by genetic mutations that affect the production or function of the glycoprotein Ib-IX-V (GPIb-IX-V) complex. This complex is located on the surface of platelets and is essential for their ability to adhere to damaged blood vessels, a critical step in the blood clotting process.
The GPIb-IX-V complex is made up of four different proteins:
- Glycoprotein Ib alpha (GP1BA)
- Glycoprotein Ib beta (GP1BB)
- Glycoprotein IX (GP9)
- Glycoprotein V (GPV)
Mutations in the genes that provide the instructions for making these proteins (GP1BA, GP1BB, or GP9) can lead to Bernard-Soulier syndrome. These mutations can result in:
- A reduced number of GPIb-IX-V complexes on the platelet surface
- The production of a dysfunctional GPIb-IX-V complex that cannot properly bind to von Willebrand factor (vWF)
In either case, the ability of platelets to adhere to damaged blood vessels is impaired, leading to the characteristic bleeding problems associated with Bernard-Soulier syndrome.
Bernard-Soulier syndrome is most commonly inherited in an autosomal recessive pattern, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the disorder. In rare cases, it can be inherited in an autosomal dominant pattern, where only one copy of the mutated gene is sufficient to cause the syndrome.
Pathophysiology of Bernard-Soulier Syndrome
The pathophysiology of Bernard-Soulier syndrome centers on the critical role of the GPIb-IX-V complex in primary hemostasis, the initial phase of blood clot formation.
Normal Platelet Adhesion
When a blood vessel is injured, the subendothelial collagen and von Willebrand factor (vWF) are exposed. In normal hemostasis, the GPIb-IX-V complex on the surface of platelets binds to vWF. This binding is essential for the initial adhesion of platelets to the site of injury, especially under high shear stress conditions (rapid blood flow).
This initial adhesion is crucial for the subsequent recruitment and activation of more platelets, leading to the formation of a stable platelet plug.
Pathophysiology in Bernard-Soulier Syndrome
In Bernard-Soulier syndrome, due to a deficiency or dysfunction of the GPIb-IX-V complex, this initial adhesion to vWF is impaired. Platelets are unable to effectively bind to the subendothelium, which disrupts the normal process of platelet plug formation.
As a result, despite the presence of other coagulation factors, the initial step of platelet adhesion is compromised, leading to a prolonged bleeding time and an increased susceptibility to bleeding.
The macrothrombocytopenia (large platelets, low platelet count) seen in BSS may be a compensatory mechanism, but the larger platelets are still functionally defective due to the GPIb-IX-V abnormality. The exact mechanism of macrothrombocytopenia in BSS is not fully understood, but it may relate to abnormal platelet production or release from megakaryocytes.
Signs and Symptoms of Bernard-Soulier Syndrome
Bernard-Soulier syndrome often presents in infancy or early childhood with bleeding symptoms. These early manifestations may include easy bruising, frequent nosebleeds, and gum bleeding. The early onset of these symptoms is due to the congenital nature of the disorder, as individuals are born with the genetic mutations that cause the GPIb-IX-V complex deficiency or dysfunction.
Common signs and symptoms include:
- Easy bruising: Individuals with Bernard-Soulier syndrome may bruise easily and the bruises may be larger or take longer to heal than in healthy individuals.
- Nosebleeds (epistaxis): Frequent or severe nosebleeds are common.
- Gum bleeding: Especially after dental procedures.
- Heavy or prolonged menstrual bleeding (menorrhagia): Can sometimes lead to iron deficiency anemia.
- Prolonged bleeding after injuries, surgery, or dental work: Even minor cuts or surgical procedures can result in excessive bleeding.
- Skin bleeding: Small red or purple spots on the skin (petechiae) or larger areas of bleeding under the skin (purpura) may occur.
- Gastrointestinal bleeding: Although less common, bleeding can occur in the stomach or intestines, leading to blood in the stool or vomit.
- Other bleeding: Rarely, bleeding may occur in other areas, such as the urinary tract (hematuria) or the brain (intracranial hemorrhage).
The severity of bleeding in Bernard-Soulier syndrome can range from mild to severe. Some individuals may only experience mild bruising or occasional nosebleeds, while others may have more frequent and severe bleeding episodes that require medical intervention, such as blood transfusions. This variability is influenced by several factors, including the specific genetic mutation, the degree of GPIb-IX-V complex deficiency or dysfunction, and individual patient factors.
While patients with Bernard-Soulier syndrome have thrombocytopenia (low platelet count), the severity of bleeding may not always directly correlate with the platelet count. This is because the functional defect in the platelets (due to the GPIb-IX-V abnormality) also plays a significant role in the bleeding tendency.
Laboratory Investigations in Bernard-Soulier Syndrome
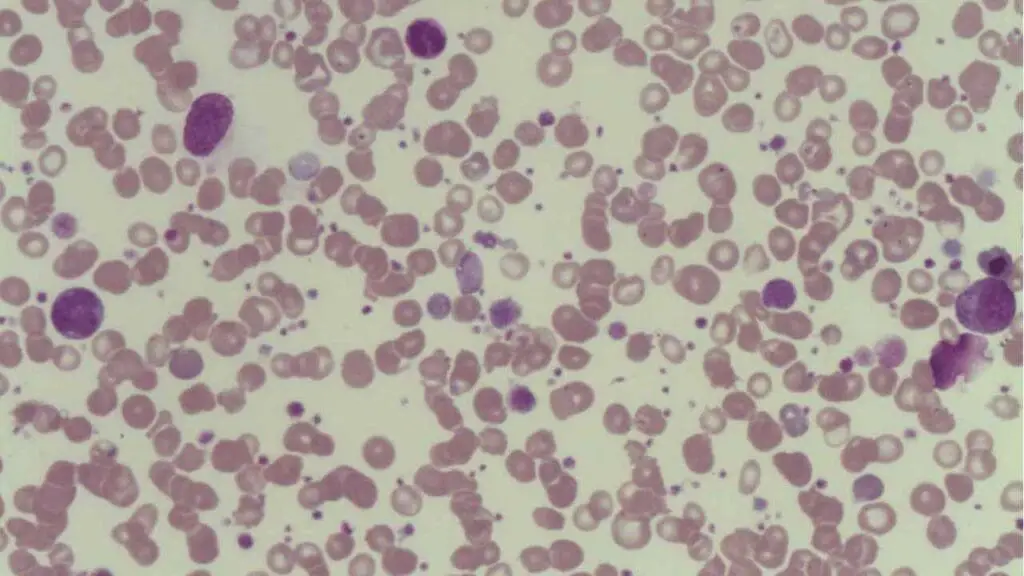
The diagnosis of Bernard-Soulier syndrome typically involves a combination of clinical evaluation and laboratory testing.
- Complete Blood Count (CBC): A low platelet count (thrombocytopenia) is a characteristic finding. Platelet counts typically range from 20 to 100 x 109/L (normal range is 150 - 450 x 109/L), but can be even lower in some cases.
- Peripheral Blood Smear: Microscopic examination of a blood smear reveals the presence of enlarged platelets, often described as "giant" platelets (macrothrombocytopenia).
- Bleeding Time: In Bernard-Soulier syndrome, the bleeding time is typically prolonged, reflecting the impaired platelet function.
- Platelet Function Tests:
- Ristocetin-induced platelet aggregation: Platelet aggregation in response to ristocetin is characteristically absent or markedly reduced. This is a crucial diagnostic test for Bernard-Soulier syndrome, as it reflects the deficiency or dysfunction of the GPIb-IX-V complex.
- Other platelet agonists: Platelet aggregation responses to other agonists such as ADP, collagen, and epinephrine are typically normal. This helps to differentiate Bernard-Soulier syndrome from other platelet function disorders like Glanzmann thrombasthenia.
- Flow Cytometry: A significant reduction or absence of one or more components of the GPIb-IX-V complex (usually GPIbα). This is a highly specific test for Bernard-Soulier syndrome.
- Genetic Testing: Genetic testing to identify mutations in the GP1BA, GP1BB, or GP9 genes can confirm the diagnosis of Bernard-Soulier syndrome and can be helpful for family studies and genetic counseling.
Differential Diagnosis of Bernard-Soulier Syndrome
Okay, here's a table presenting the differential diagnosis of Bernard-Soulier Syndrome:
| Condition | Low Platelet Count | Giant Platelets | Ristocetin-induced Aggregation | Other Key Features |
| Bernard-Soulier Syndrome (BSS) | ✔ | ✔ | Absent/Markedly Reduced | Giant platelets on smear; decreased GPIb-IX-V on flow cytometry |
| Immune Thrombocytopenia (ITP) | ✔ | ❌ | Normal | Normal or small platelets; autoimmune etiology |
| von Willebrand Disease (vWD) | ❌ | ❌ | Abnormal, corrected by plasma | Variable vWF levels; abnormal vWF function tests |
| Glanzmann Thrombasthenia | ❌ | ❌ | Normal | Normal platelet count and morphology; absent aggregation to ADP, collagen, epinephrine |
| Gray Platelet Syndrome (GPS) | Mild | ✔ | Normal or reduced | Large, pale platelets; deficiency of alpha-granules |
| May-Hegglin Anomaly | ✔ | ✔ | Normal | Döhle bodies in leukocytes; variable bleeding tendency |
Treatment and Management
The treatment and management of Bernard-Soulier Syndrome focus on preventing and controlling bleeding episodes, as there is no cure for the underlying genetic defect.
General Principles
- Multidisciplinary care: Hematologists, dentists, and other specialists collaborate.
- Patient education: Crucial for understanding the condition and preventive measures.
- Avoidance of aggravating factors: Drugs that impair platelet function (e.g., aspirin, NSAIDs) should be avoided.
Management of Acute Bleeding
- Local hemostatic measures: Applying pressure to the bleeding site or use topical hemostatic agents (e.g., fibrin sealants)
- Platelet transfusions: It is effective for significant bleeding. However, there is a risk of alloimmunization (development of antibodies against donor platelets) with repeated transfusions, making future transfusions less effective. The use of HLA-matched platelets can reduce alloimmunization risk.
- Recombinant activated factor VIIa (rFVIIa): May be used in cases of severe bleeding or when platelet transfusions are ineffective or contraindicated.
Prophylactic Measures
- Antifibrinolytic agents: Medications like tranexamic acid can reduce bleeding, especially mucosal bleeding (e.g., nosebleeds, menorrhagia).
- Careful management of procedures: Minimizing trauma during surgery, dental work, and other invasive procedures. Prophylactic platelet transfusions or rFVIIa may be considered before major surgery.
Specific Considerations
- Menorrhagia: Antifibrinolytic agents and hormonal therapies
- Pregnancy and delivery: Close monitoring and planning. Platelet transfusions may be necessary during delivery.
- Dental procedures: Local hemostatic measures, antifibrinolytics. Platelet transfusions or rFVIIa may be needed for extensive procedures.
Prognosis
The long-term prognosis depends on the severity of the bleeding tendency and the effectiveness of management strategies. With proper management, most individuals with Bernard-Soulier syndrome can lead relatively normal lives.
However, the risk of bleeding complications, especially in cases of severe BSS, remains a concern. Complications related to treatment, such as alloimmunization from platelet transfusions, can also affect the prognosis.
Frequently Asked Questions (FAQs)
What is the life expectancy of someone with Bernard-Soulier syndrome?
With proper care and management, Bernard-Soulier Syndrome (BSS) typically does not reduce life expectancy. Most individuals with BSS can live full lives by taking precautions, receiving prompt treatment for bleeding episodes, and having regular medical follow-up. Although the severity of bleeding varies among individuals, those with both mild and severe forms of the condition can often achieve a good long-term outcome with appropriate care.
Which characteristic of Bernard-Soulier syndrome helps distinguish it from von Willebrand disease?
Bernard-Soulier syndrome (BSS) and von Willebrand disease (VWD) are both bleeding disorders, but they differ in key characteristics. While both can cause prolonged bleeding time, the presence of abnormally large platelets is a hallmark of BSS and is typically not seen in VWD. Additionally, while ristocetin-induced platelet aggregation is abnormal in both, it can be corrected by adding normal plasma in VWD, but not in BSS.
Why are platelets enlarged in Bernard Soulier?
The reason platelets are enlarged in Bernard-Soulier Syndrome is due to a defect in the glycoprotein Ib-IX-V complex, which is crucial for platelet formation and regulation of their size. This defect disrupts the normal process of platelet production in the bone marrow, leading to the release of abnormally large platelets into the circulation.
What is the age of presentation of Bernard-Soulier syndrome?
Bernard-Soulier syndrome typically presents in infancy or early childhood as it is a genetic disorder.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Goldberg S, Hoffman J. Clinical Hematology Made Ridiculously Simple, 1st Edition: An Incredibly Easy Way to Learn for Medical, Nursing, PA Students, and General Practitioners (MedMaster Medical Books). 2021.
- Almomani MH, Mangla A. Bernard-Soulier Syndrome. [Updated 2024 Jan 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557671/
- Berndt MC, Andrews RK. Bernard-Soulier syndrome. Haematologica. 2011 Mar;96(3):355-9. doi: 10.3324/haematol.2010.039883. PMID: 21357716; PMCID: PMC3046265.