Key Takeaways
Hemophilia A (Classic Hemophilia) is an inherited bleeding disorder defined by a quantitative or qualitative deficiency of Factor VIII (FVIII).
- Etiology & Inheritance ▾: Hemophilia A is an X-linked recessive disorder caused by mutations in the F8 gene on the X chromosome. It affects males almost exclusively, while females are typically asymptomatic carriers. The most common severe mutation is the Intron 22 Inversion.
- Pathophysiology ▾: The lack of functional FVIII impairs the formation of the Tenase Complex (FVIIIa + FIXa) in the intrinsic pathway of the coagulation cascade, leading to a failure of stable fibrin clot formation.
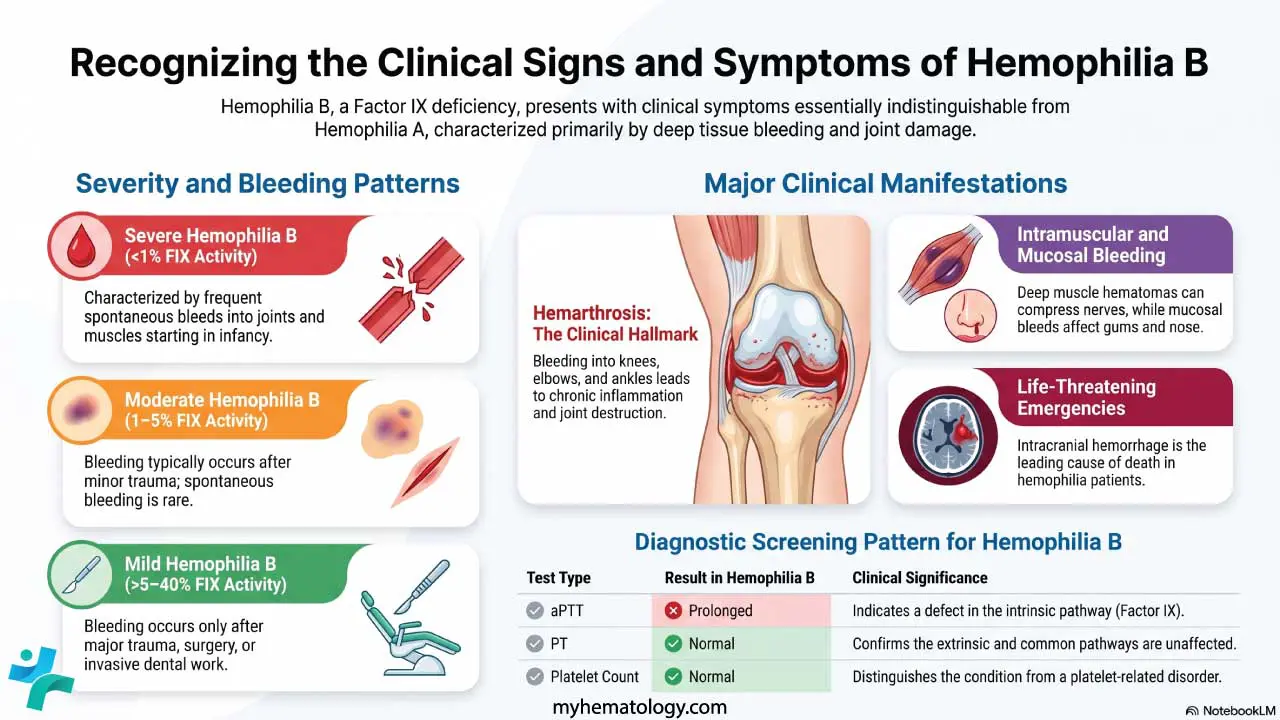
- Clinical Hallmark ▾: The characteristic manifestation of Hemophilia A is deep, spontaneous bleeding, particularly hemarthrosis (bleeding into joints, most commonly knees, elbows, and ankles) and intramuscular hematomas.
- Severity Classification: Clinical severity of Hemophilia A is directly tied to FVIII activity:
- Severe (< 1% activity): Characterized by frequent spontaneous bleeds.
- Moderate (1–5% activity): Bleeds only after minor trauma.
- Mild (> 5%–40% activity): Bleeds only after major trauma or surgery.
- Diagnosis ▾: Screening tests in Hemophilia A show a characteristically prolonged activated Partial Thromboplastin Time (aPTT), while Prothrombin Time (PT), bleeding time, and platelet count are typically normal. Diagnosis is confirmed by a low FVIII functional assay.
- Treatment & Management ▾: Management of Hemophilia A relies on prophylaxis (routine infusions of recombinant FVIII or Factor VIII-sparing agents like Emicizumab) to maintain factor levels above 1% and prevent joint damage.
- Complications: The most serious treatment complication of Hemophilia A is the development of inhibitors (neutralizing antibodies) against infused FVIII, which renders standard replacement therapy ineffective.
*Click ▾ for more information
Introduction
Hemophilia A, often referred to as Classic Hemophilia, is a severe, lifelong, inherited bleeding disorder characterized by a quantitative deficiency or a functional defect of Coagulation Factor VIII (FVIII). This congenital clotting factor deficiency impairs the body’s ability to form a stable fibrin clot, resulting in recurrent and prolonged bleeding episodes.
Epidemiology
Hemophilia A is the most common severe inherited bleeding disorder worldwide, affecting approximately 1 in 5,000 male live births. Because the gene for FVIII (F8) is located on the X chromosome, the condition of Hemophilia A follows an X-linked recessive inheritance pattern, meaning it primarily affects males. While females can be symptomatic carriers, the vast majority of severe cases occur in men. Hemophilia A accounts for roughly 80% to 85% of all hemophilia cases, making it significantly more prevalent than Hemophilia B (Factor IX deficiency).

Historical Context
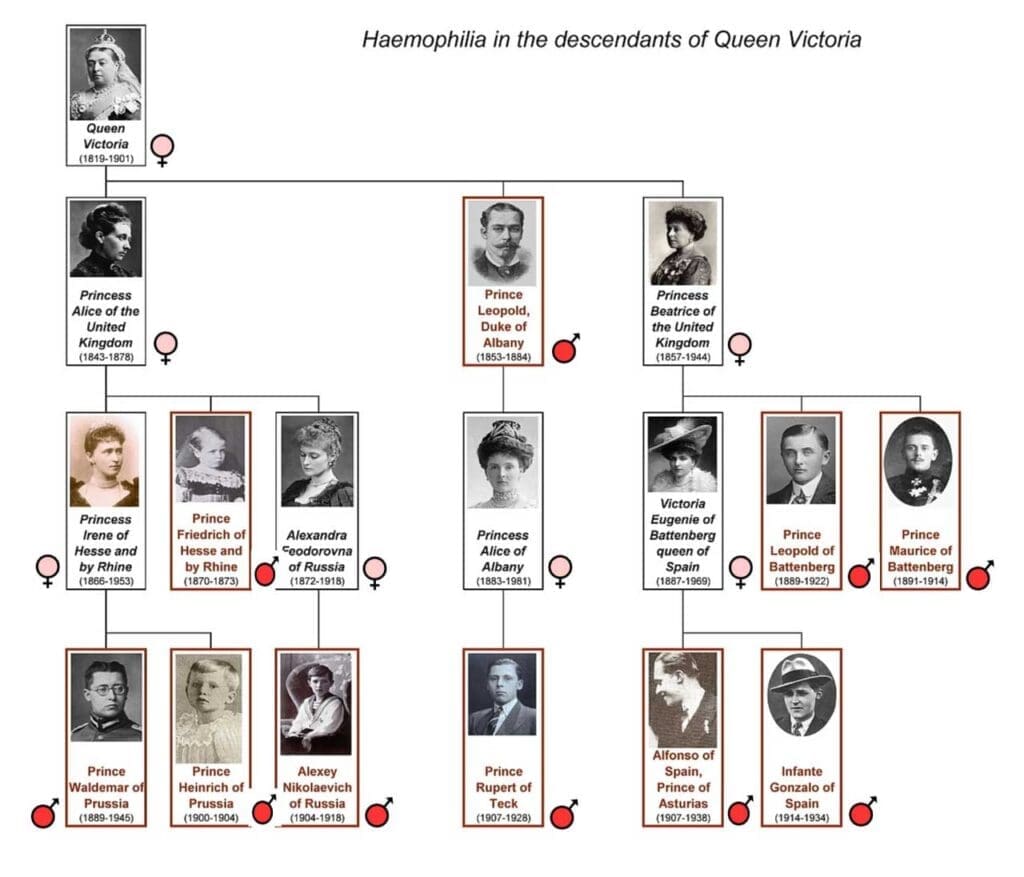
Due to its distinctive X-linked inheritance pattern, hemophilia has been recognized clinically for centuries, first appearing in the Babylonian Talmud in the 2nd century AD. However, it achieved global notoriety during the 19th and 20th centuries as "the Royal Disease."
This designation stems from the fact that a mutation in the F8 gene arose spontaneously in Queen Victoria of England or her immediate ancestors. Since she was a carrier, the gene was passed down through her children to the royal houses of Germany, Spain, and, most famously, Russia, severely affecting individuals like Tsarevich Alexei. This historical case provides a compelling real-world illustration of X-linked recessive inheritance in a well-documented lineage.
Molecular and Cellular Pathophysiology
The Role of Factor VIII
Factor VIII is a large, essential glycoprotein synthesized primarily in the liver sinusoidal cells and endothelium. In the circulation, FVIII is non-covalently bound to von Willebrand Factor (vWF), which stabilizes and protects it from premature clearance and degradation.
FVIII itself is not an enzyme. It is a cofactor, meaning it is a non-protein chemical compound that is required for a protein's biological activity. Specifically, FVIII acts as a powerful enhancer for Factor IXa (FIXa).
In its inactive circulating form, FVIII is non-covalently bound to von Willebrand Factor (vWF). This complex serves two vital purposes:
- Protection: vWF shields FVIII from rapid degradation by activated protein C, significantly extending its half-life in the circulation.
- Transport: vWF serves as the carrier protein, transporting FVIII to the site of vascular injury.
Upon injury, the coagulation cascade is triggered, and a small amount of Thrombin (Factor IIa) is generated. This initial thrombin then cleaves FVIII from vWF, releasing the FVIII molecule and activating it into its highly potent form, FVIIIa.
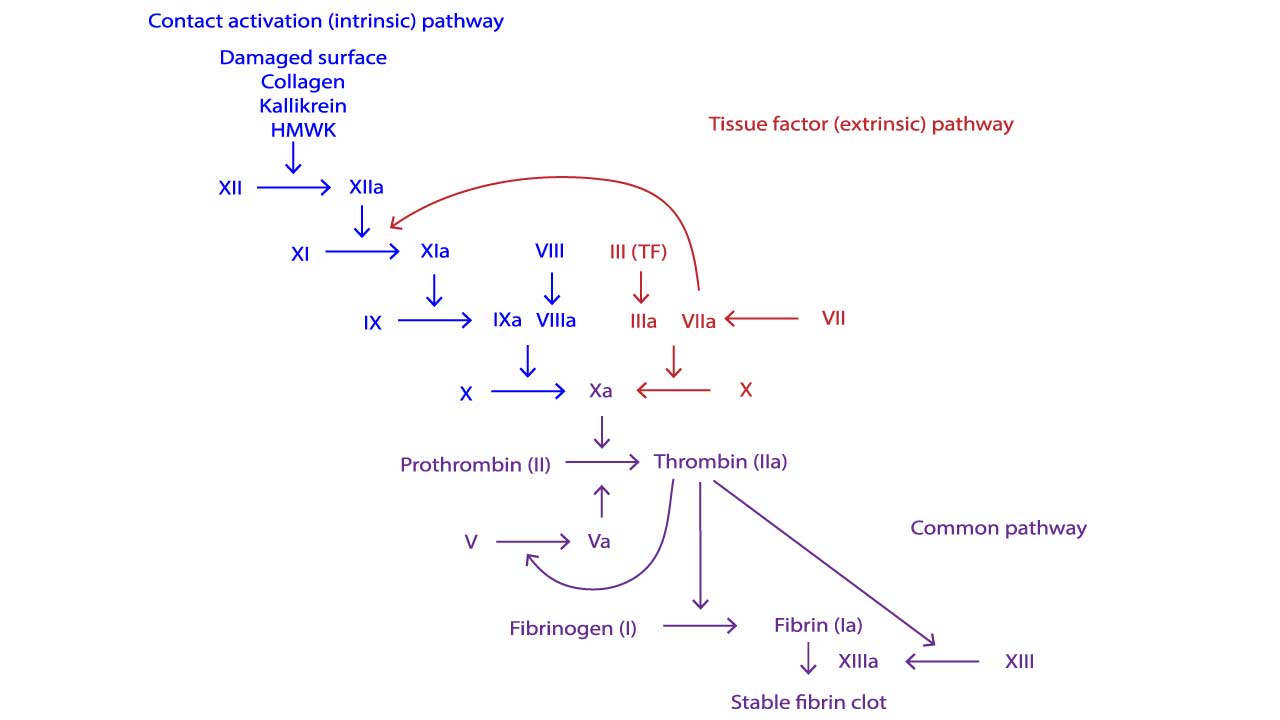
The Intrinsic Tenase Complex
The central role of FVIIIa is in forming the intrinsic tenase complex (also known as the Factor X-activating complex). This complex is the engine of the intrinsic pathway.The intrinsic tenase complex is composed of four essential elements, assembled on a phospholipid surface (provided by activated platelets):
- Enzyme: Activated Factor IX (FIXa)
- Substrate: Factor X (FX)
- Cofactor: Activated Factor VIII (FVIIIa)
- Mineral: Calcium ions (Ca2+)
When FVIII is activated to FVIIIa, it binds FIXa and positions it optimally next to the substrate FX. FVIIIa increases the catalytic efficiency of FIXa by up to 200,000-fold.
The rapid activation of FX to FXa is paramount because FXa is the first enzyme of the common pathway, leading immediately to the formation of the Prothrombinase Complex and the critical thrombin burst.
The Pathophysiological Defect
In Hemophilia A, the deficient or dysfunctional FVIII causes a major defect in the intrinsic tenase complex formation. Without sufficient FVIIIa, FIXa can only activate FX at an extremely slow, basal rate. This prevents the rapid, explosive generation of thrombin (the thrombin burst) that is necessary to form a large, stable, cross-linked fibrin clot.
While the initial platelet plug forms (primary hemostasis is intact), the resultant fibrin network in Hemophilia A is too weak, too slow to form, and insufficient to withstand the pressure of blood flow. This defect in stabilizing the clot leads to the characteristic deep tissue and joint bleeding (hemarthrosis) seen in hemophilia patients, where mechanical stress requires a rapid and robust fibrin seal.
The Genetic Basis of Hemophilia A
Hemophilia A is caused by mutations in the F8 gene, located on the long arm of the X chromosome. Two copies of the mutated gene are required for the condition to manifest in females (XX), while only one copy is needed for males (XY). Since males have only one X chromosome, they are always affected if they inherit the mutated gene.
Females who possess one normal F8 gene and one mutant F8 gene are typically asymptomatic carriers, as the normal X chromosome usually produces enough Factor VIII to maintain adequate hemostasis. However, due to the phenomenon of X-chromosome inactivation (Lyonization), a small percentage of female carriers may have factor levels low enough to exhibit mild or, rarely, moderate symptoms.
The F8 Gene
The genetic defect is localized to the Factor VIII gene (F8), which is one of the largest genes in the human genome, spanning 186 kilobases (kb) of genomic DNA and containing 26 exons.
The F8 gene is situated at the terminal end of the long arm of the X chromosome, specifically at locus Xq28. It encodes a large glycoprotein precursor that is cleaved and secreted into the bloodstream. This large size makes the gene highly susceptible to various mutations.
Common Mutations Leading to Hemophilia A
The wide range of Factor VIII activity seen clinically (severe, moderate, mild) directly correlates with the type and location of the mutation in the F8 gene.
- Intron 22 Inversion (I22Inv) This is the most common single molecular defect, responsible for approximately 40–50% of all cases of severe Hemophilia A. The inversion involves a large portion of the gene and results in a truncated, non-functional FVIII protein. Because the protein is completely non-functional or absent, this mutation almost exclusively causes the severe phenotype (FVIII activity < 1%).
- Intron 1 Inversion (I1Inv): A less common, but still clinically relevant, inversion affecting intron 1. It is responsible for 2–5% of severe cases.
- Point Mutations (Missense, Nonsense): These involve a single base pair change in the gene sequence. They typically result in the mild or moderate forms of hemophilia A, as the gene may still produce some residual, partially functional FVIII protein, or a reduced quantity of normal protein.
- Large Deletions and Insertions: These structural mutations are less common but often lead to the severe phenotype due to the complete absence of FVIII synthesis. Notably, large deletions are often associated with a higher risk of inhibitor development because the patient's immune system has no baseline exposure to any part of the FVIII protein.
Clinical Manifestation of Hemophilia A
The clinical presentation of Hemophilia A is highly variable, but it follows a predictable pattern dictated almost entirely by the residual functional plasma activity of Factor VIII (FVIII). The lower the factor level, the more frequent, severe, and spontaneous the bleeding episodes will be.
Severity Correlation
Hemophilia A is classified into three clinically distinct categories based on FVIII activity levels. This classification dictates the patient's lifelong bleeding risk and, consequently, the primary treatment strategy (prophylaxis vs. on-demand treatment).
| Severity Level | FVIII Plasma Activity (% of Normal) | Clinical Presentation |
| Severe | < 1% | Frequent, spontaneous bleeding into joints and muscles (2–5 episodes per month). Bleeding begins in infancy/toddler years as the child becomes mobile. |
| Moderate | 1% – 5% | Bleeding only occurs after minor trauma or invasive procedures. Spontaneous bleeds are rare. May be diagnosed later in childhood. |
| Mild | > 5% – 40% | Bleeding occurs only after major trauma, surgery, or significant dental work. Many individuals may not be diagnosed until adulthood. |
Key Hemorrhagic Manifestations
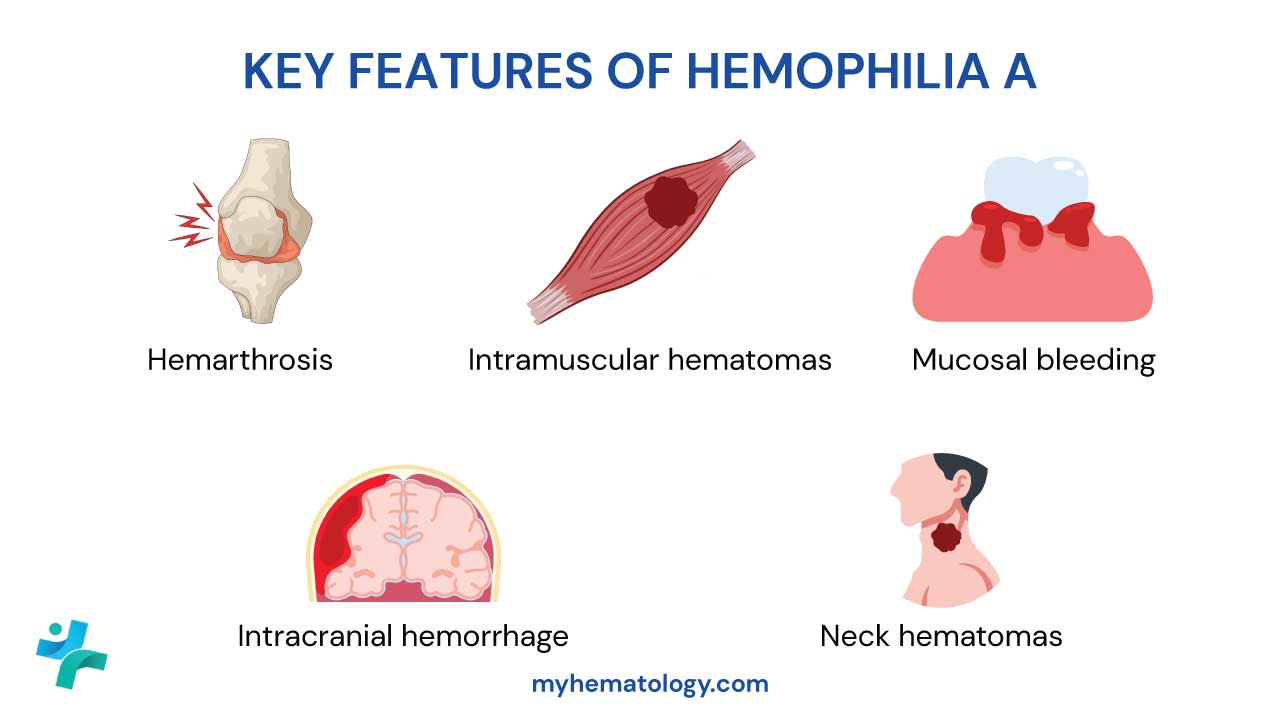
The pattern of bleeding in hemophilia is distinct from platelet disorders. The defect in secondary hemostasis leads to delayed, deep, and often recurrent bleeding episodes.
Hemarthrosis (Joint Bleeds)
This is the hallmark of hemophilia and the primary cause of long-term disability. Bleeding into the joint space (synovial capsule) is typically spontaneous in severe hemophilia. The most commonly affected target joints are the knees, elbows, and ankles (in that order), followed by the hips and shoulders.
- Acute Signs: The initial symptoms include a painful, stiff, and warm joint, often preceded by a tingling sensation (an aura) recognized by the patient. If untreated, the joint swells due to blood accumulation.
- Hemophilic Arthropathy (Chronic Damage): Recurrent hemarthrosis leads to chronic inflammation (synovitis), destruction of cartilage and bone, and ultimately, irreversible joint damage, contractures, and functional disability. This is the definition of hemophilic arthropathy.
Intramuscular Hematomas
Bleeding into deep muscle tissue is common, particularly after a fall or minor trauma. Large hematomas can cause nerve compression (e.g., femoral nerve compression from an iliopsoas bleed) or, critically, compartment syndrome, which requires urgent Factor VIII replacement and potentially surgical intervention.
Mucosal Bleeding
While less frequent than joint bleeds, mucosal bleeding can be significant:
- Gingival Bleeding: Prolonged bleeding after tooth brushing or dental procedures.
- Epistaxis: Difficult-to-control nosebleeds.
- Gastrointestinal (GI) Bleeding: Less common but potentially serious, leading to chronic iron-deficiency anemia or acute hemorrhage.
Life-Threatening Hemorrhages
These require immediate and aggressive Factor VIII replacement therapy, aiming for 100% factor activity levels.
- Intracranial Hemorrhage (ICH): This is the most common cause of death in hemophilia patients. It can be spontaneous, secondary to minor trauma, or even occur perinatally. Signs include persistent headache, vomiting, and altered consciousness.
- Oropharyngeal/Neck Hematomas: Bleeding in the soft tissues of the throat or neck can quickly compress the airway, leading to obstruction and asphyxiation.
Bleeding in Neonates
Hemophilia A may be suspected at birth if there is a family history. Neonatal presentations include:
- Cephalohematoma: Bleeding under the scalp.
- Intracranial Hemorrhage (ICH): Can occur during a traumatic delivery.
- Excessive Bleeding: Following circumcision (if performed before diagnosis and treatment).
Diagnosis and Monitoring
The diagnosis of Hemophilia A is typically initiated by a patient history revealing characteristic bleeding symptoms (often hemarthrosis) or a known family history. However, confirmation requires a sequential process involving screening tests and definitive functional assays.
Initial Screening Tests
Initial assessment relies on the standard battery of coagulation tests. In Hemophilia A, the results show a characteristic pattern pointing directly to a defect in the intrinsic pathway.
| Test | Expected Results | Rationale |
| Activated Partial Thromboplastin Time (aPTT) | Prolonged | The aPTT specifically measures the time required for plasma to clot via the intrinsic and common pathways. Since Factor VIII is essential for the intrinsic pathway, its deficiency significantly extends the clotting time. |
| Prothrombin Time (PT) | Normal | The PT measures the extrinsic and common pathways. Since Factor VII (extrinsic) and the common pathway factors (X, V, II, I) are unaffected, this test remains normal. |
| Thrombin Time (TT) | Normal | Measures the final step of clot formation: fibrinogen > fibrin. Since fibrinogen and thrombin are normal, the TT is typically normal. |
| Platelet Count | Normal | Hemophilia is a defect of plasma clotting factors, not platelets. Platelet count and function are usually normal. |
Confirmatory and Quantitative Testing
While a prolonged aPTT with a normal PT is highly suggestive of a factor deficiency, the definitive diagnosis and severity classification rely on specific Factor VIII assays.
Specific Functional Factor VIII (FVIII:C) Activity Assay
This is the gold standard for confirming the diagnosis and determining severity. This test measures the actual ability of the patient's plasma to correct the clotting time of Factor VIII-deficient plasma. The result is expressed as a percentage of normal pooled plasma activity.
The measured activity level directly classifies the patient's severity (Severe: <1%; Moderate: 1–5%; Mild: 5–40%).
Factor VIII Antigen (FVIII:Ag) Level
This assay measures the amount of Factor VIII protein present, regardless of whether it is functional. In most cases of Hemophilia A, both FVIII:C (activity) and FVIII:Ag (amount) are concordantly low. However, a select group of patients may produce an abnormally structured, non-functional FVIII protein (a qualitative defect). In these rare cases, the FVIII:Ag level may be normal or near-normal, while the FVIII:C is low.
Genetic and Molecular Testing
Genetic testing is crucial for defining the specific mutation in Hemophilia A, which has implications for prognosis and inhibitor risk, and for screening family members.
Molecular testing identifies the specific mutation in the F8 gene (e.g., Intron 22 Inversion). This information is valuable because certain mutations (like large deletions or the I22 inversion) are strongly correlated with a high risk of inhibitor development.
Differential Diagnosis
The key differential diagnosis for a patient presenting with a prolonged aPTT and normal PT is:
- Hemophilia B (Factor IX Deficiency): Clinically indistinguishable, but confirmed by a low Factor IX activity level and a normal FVIII activity level.
- Severe Type 3 von Willebrand Disease (vWD): This is characterized by extremely low levels of von Willebrand Factor (vWF). Since vWF protects FVIII, a severe vWF deficiency will lead to a secondary Factor VIII deficiency, mimicking Hemophilia A. Differentiation is achieved by measuring vWF:Ag and vWF activity (which will be low in vWD, but normal in Hemophilia A).
Clinical Management
The primary goal of Hemophilia A management is to prevent joint damage and chronic disability, and to promptly treat acute bleeding episodes. The standard of care has evolved dramatically from reactive treatment to proactive prevention, leading to vastly improved quality of life and life expectancy for patients.
Factor VIII Replacement Therapy: The Gold Standard
Treatment relies on increasing the deficient Factor VIII (FVIII) activity level in the plasma.
Factor VIII Products
Modern treatment almost exclusively uses Recombinant Factor VIII (rFVIII) products.
- Recombinant FVIII (rFVIII): Genetically engineered and produced outside the human blood supply, eliminating the risk of transmitting blood-borne viruses (e.g., HIV, HCV).
- Extended Half-Life (EHL) FVIII: Modified FVIII molecules (often pegylated or Fc-fusion proteins) designed to stay active in the bloodstream longer, reducing the required frequency of intravenous (IV) injections, thus improving patient adherence.
Treatment Strategies
| Strategy | Goal | Indication | Frequency |
| Prophylaxis (Preventative) | Maintain trough FVIII activity levels above 1–2% to prevent spontaneous bleeds. | Severe Hemophilia A. Also often used for moderate patients with a history of recurrent bleeding (target joints). | Typically 2–3 times per week (or less with EHL products) via IV infusion. |
| On-Demand (Reactive) | Treat bleeding only when it occurs. | Mild Hemophilia A or occasional use in moderate patients. | Administered at the time of a bleed until the bleeding stops. |
| Surgical Prophylaxis | Maintain FVIII levels at 80–100% pre-procedure and 50–80% post-procedure. | All major surgeries or invasive dental procedures. | Pre-operative dose, followed by sustained dosing for the healing period. |
Treatment of Acute Bleeds
Prompt administration of FVIII concentrate is essential to minimize damage, particularly during a hemarthrosis (joint bleed). Dosing is calculated based on the patient's weight and the desired target level, typically aiming for 50–100% activity, depending on the severity of the bleed.
Acute Management: The R.I.C.E. principle is applied alongside factor infusion:
- Rest: Immobilize the affected joint.
- Ice: Apply cold packs to reduce swelling.
- Compression: Gently wrap the area.
- Elevation: Raise the limb to reduce blood flow and swelling.
Development of Factor VIII Inhibitors
The development of inhibitors (alloantibodies) against infused FVIII is the most serious complication of treatment, occurring in approximately 25–30% of severe Hemophilia A patients. These antibodies neutralize the therapeutic Factor VIII, rendering standard replacement therapy ineffective.
Detection and Bypassing Agents
Inhibitor presence is confirmed using the Bethesda Assay, which quantifies the inhibitor strength in Bethesda Units (BU).
When inhibitors are present, treatment shifts to agents that bypass the FVIII/FIX step and activate the common pathway directly:
- Activated Prothrombin Complex Concentrate (aPCC): Contains activated clotting factors (II, VIIa, IXa, Xa).
- Recombinant Factor VIIa (rFVIIa): Binds to tissue factor, rapidly activating the extrinsic pathway to generate thrombin.
Immune Tolerance Induction (ITI)
ITI is the definitive strategy to eradicate inhibitors. High, frequent doses of Factor VIII concentrate are infused, overwhelming the immune system and training it to tolerate the Factor VIII protein. ITI is highly successful (around 70–80% success rate), particularly when started early.
Novel Non-Factor Replacement Therapies
These agents are revolutionizing care, especially for patients with inhibitors, by replacing the function of FVIII without being susceptible to anti-FVIII antibodies.
- Emicizumab (Hemlibra): Emicizumab is a bispecific monoclonal antibody that bridges Factor IXa and Factor X, effectively mimicking the cofactor function of FVIIIa. Approved for prophylaxis in both patients with and without FVIII inhibitors, fundamentally changing the treatment landscape by offering highly effective, non-IV prophylaxis.
- Emerging Therapies
- Fitusiran: An RNA interference (RNAi) therapeutic that lowers the levels of Antithrombin, a natural anticoagulant. Lowering antithrombin effectively pushes the coagulation balance toward clotting, compensating for the FVIII deficiency.
- Concizumab: A monoclonal antibody that blocks the anticoagulant function of Tissue Factor Pathway Inhibitor (TFPI).
Adjunctive Therapies
- Desmopressin (DDAVP): A synthetic analog of vasopressin. DDAVP causes the release of stored FVIII and vWF from endothelial cells. However, only effective for patients with Mild Hemophilia A who can produce some FVIII. It is ineffective in moderate or severe patients.
- Antifibrinolytics (e.g., Tranexamic Acid): Used to stabilize clots in areas rich in fibrinolytic enzymes, such as the mouth (dental procedures) and mucosa.
Complications and Long-Term Sequelae of Hemophilia A
While modern prophylactic treatment has drastically reduced morbidity and mortality, individuals with Hemophilia A still face significant long-term health challenges. These complications fall into two main categories: those arising directly from recurrent bleeding and those arising from treatment.
Hemophilic Arthropathy (Chronic Joint Disease)
This is the most common and devastating complication of Hemophilia A, leading to progressive disability and reduced quality of life.
The Cycle of Joint Destruction
- Acute Hemarthrosis: Blood accumulates in the joint space (most commonly knees, elbows, ankles).
- Synovitis: The presence of iron (from broken-down red blood cells) in the joint space is highly toxic. It triggers a chronic inflammatory response in the synovial membrane (the lining of the joint).
- Synovial Hypertrophy: The inflamed synovium thickens, develops new fragile blood vessels, and produces destructive enzymes (angiogenesis). This state is known as chronic synovitis.
- Target Joint Formation: The fragile, hypervascular synovium is highly prone to re-bleeding, creating a vicious cycle. Joints that experience three or more bleeds within a six-month period are termed target joints.
- Cartilage and Bone Destruction: The destructive enzymes released by the thickened synovium break down articular cartilage and subchondral bone, resulting in permanent joint damage, pain, stiffness, muscle atrophy, and eventual requirement for joint replacement surgery.
Management of Arthropathy
Management is primarily preventative (prophylaxis) but also involves surgical and rehabilitative measures.
- Synovectomy: Surgical removal of the damaged, hyperplastic synovium (either chemically, radioactively, or surgically) to break the cycle of re-bleeding.
- Physiotherapy: Essential for maintaining muscle strength, joint range of motion, and stability, slowing the progression of damage.
- Joint Replacement: For end-stage arthropathy, total joint replacement (e.g., knee or hip) is required to restore function.
Factor VIII Inhibitors
As detailed in the treatment section, the development of neutralizing alloantibodies (inhibitors) against Factor VIII is the most serious treatment complication.
Risk Factors
Inhibitor development is an immunological complication strongly associated with:
- Severity: Almost exclusively occurs in patients with Severe Hemophilia A.
- Genetics: Patients with large deletions or the Intron 22 Inversion mutation in the F8 gene have a higher risk.
- Exposure: Most inhibitors develop early in life, within the first 50 exposure days to Factor VIII products.
An inhibitor turns a severe, but treatable, case of Hemophilia A into an extremely complex and expensive condition. The standard Factor VIII prophylaxis becomes ineffective, requiring the reliance on Bypassing Agents (aPCC, rFVIIa) or novel non-factor therapies like Emicizumab.
Other Long-Term Issues
- Bone Health (Osteoporosis/Osteopenia): Chronic joint inflammation, limited mobility, and the inflammatory process itself contribute to reduced bone mineral density, increasing the risk of pathological fractures.
- Central Venous Access Device (CVAD) Complications: Patients on long-term prophylaxis often require surgically implanted ports for IV access. These carry risks of infection, thrombosis, and mechanical failure.
- Viral Infections: Although largely eliminated with modern recombinant factor products, historical cohorts of hemophilia patients who received plasma-derived products before viral inactivation techniques were common still suffer from chronic Hepatitis C (HCV) and HIV/AIDS infection. These patients require specialized co-management.
- Quality of Life and Psychological Burden: The continuous need for infusions, chronic pain from joint damage, fear of bleeding, and the high cost/complexity of treatment impose a significant psychological burden on both the patient and their family.
Emerging Therapeutic Directions: Gene Therapy
The development of Factor VIII replacement therapy and non-factor products like Emicizumab has revolutionized hemophilia care. However, the ultimate therapeutic goal is to cure the disorder by providing patients with a sustained, endogenous source of functional Factor VIII. This goal is currently being realized through gene therapy.
Gene Therapy: The Concept
Gene therapy for hemophilia aims to introduce a functional copy of the F8 gene into the patient’s cells, enabling them to produce FVIII protein permanently. This eliminates the need for frequent, often lifelong, infusions.
Viral Vector Delivery
- Vector Choice: The primary delivery vehicle for the F8 gene is the Adeno-Associated Virus (AAV) vector. AAV is chosen because it is non-pathogenic, non-integrating (meaning it stays in the cell's nucleus without permanently altering the host genome), and highly effective at targeting the liver.
- Target Tissue: The liver is the ideal target because it is the body's natural factory for producing most clotting factors.
Mechanism of Action
- Infusion: The AAV vector carrying the functional F8 gene is infused intravenously.
- Transduction: The vector travels to the liver and enters the hepatocytes (liver cells).
- Expression: The new F8 gene (called the transgene) forms a circular piece of DNA (an episome) in the nucleus. This episome acts as a template, causing the hepatocyte to begin synthesizing and secreting functional FVIII into the bloodstream.
Clinical Status and Expectations
Gene therapy for Hemophilia A has demonstrated profound clinical efficacy in trials, moving it into clinical practice.
- Roctavian (valoctocogene roxaparvovec): This product is a critical milestone, representing the first approved gene therapy for adults with severe Hemophilia A. It uses an AAV5 vector to deliver the functional F8 gene. Its approval signifies a shift in therapeutic strategy, offering a single-infusion treatment for long-term FVIII expression.
- Sustained FVIII Levels: Patients who respond to therapy typically achieve FVIII levels in the mild range (5% to 40%). Crucially, maintaining levels above 5% virtually eliminates spontaneous bleeding, effectively converting severe hemophilia into a mild, clinical phenotype.
- Elimination of Prophylaxis: Successful gene therapy allows the majority of patients to discontinue routine Factor VIII prophylaxis and live an active, bleed-free life, only needing treatment for severe trauma or surgery.
Key Challenges and Limitations
Despite its revolutionary potential, gene therapy faces important hurdles.
- Pre-existing Immunity: Many individuals have been exposed to AAV in the general population and possess pre-existing antibodies against the AAV vector. These antibodies neutralize the infused vector, preventing the gene from reaching the liver. Patients must be screened for anti-AAV antibodies; those with high titers are currently excluded from treatment.
- Vector Safety and Toxicity (Hepatotoxicity): The body's immune system recognizes the viral vector as foreign, which often leads to a transient, asymptomatic rise in liver enzymes (transaminitis). This is usually managed effectively with a temporary course of corticosteroids.
- Durability and Longevity: As the episome (the FVIII template) does not integrate into the genome, it may slowly degrade over many years as liver cells naturally turn over. While early results show excellent long-term durability, the precise length of FVIII expression remains an active area of research.
- Re-dosing: Due to the immune response against the AAV vector, re-dosing the same vector is not currently possible.
Frequently Asked Questions (FAQs)
What is the main cause of long-term morbidity in Hemophilia A?
The main long-term morbidity is hemophilic arthropathy (joint disease). Recurrent bleeding into the joints (hemarthroses), particularly the knees, elbows, and ankles, leads to chronic synovial inflammation, cartilage destruction, and progressive loss of joint function, severely impacting quality of life.
What is the target trough FVIII level for severe Hemophilia A patients on prophylaxis?
The standard goal is to maintain a trough FVIII level of >1 IU/dL (1%). Newer studies and personalized prophylaxis (e.g., using pharmacokinetic tailoring) aim for higher troughs (e.g., 3 - 5 IU/dL) in patients with high-risk bleeding phenotypes or pre-existing arthropathy.
When is prophylactic Factor replacement typically initiated?
Primary prophylaxis is recommended for males with severe Hemophilia A before the second life-threatening joint bleed and generally before the age of 2 - 3 years, ideally as soon as the diagnosis is confirmed and venous access is established, to prevent the onset of joint damage.
What is the major drawback of Emicizumab therapy?
While effective and convenient, the major concern is the potential for thrombotic events (e.g., thrombotic microangiopathy or venous thromboembolism) when Emicizumab is co-administered with high-dose bypassing agents (FEIBA or rFVIIa) for breakthrough bleeds. Clinicians must strictly adhere to recommended bypassing agent dose limits in Emicizumab-treated patients.
Glossary of Medical Terms
- Bethesda Unit (BU): A measure of the concentration of Factor VIII inhibitor. 1 BU neutralizes 50% of the FVIII in a test plasma sample.
- Coagulation Cascade: A complex series of sequential enzyme-driven reactions leading to the formation of a fibrin clot. It involves the intrinsic, extrinsic, and common pathways.
- Factor VIII (FVIII): A glycoprotein crucial for the intrinsic pathway, serving as a cofactor for Factor IXa to activate Factor X.
- Hemarthrosis: Bleeding into a joint cavity, the hallmark manifestation of severe Hemophilia A.
- Hemophilic Arthropathy: Chronic, debilitating joint disease resulting from recurrent hemarthroses.
- Inhibitors (FVIII Alloantibodies): Antibodies produced by the immune system that neutralize the infused therapeutic Factor VIII, making replacement therapy ineffective.
- ITI (Immune Tolerance Induction): A long-term treatment strategy involving high-dose FVIII infusions aimed at eradicating FVIII inhibitors.
- Prophylaxis: Scheduled, regular administration of FVIII or non-factor therapies to prevent bleeding events.
- Trough Level: The lowest concentration of the therapeutic agent (FVIII) in the blood, measured just before the next scheduled infusion.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Nathwani AC. Gene therapy for hemophilia. Hematology Am Soc Hematol Educ Program. 2022 Dec 9;2022(1):569-578. doi: 10.1182/hematology.2022000388. PMID: 36485127; PMCID: PMC9821304.
- Brod, M., Bushnell, D. M., Busk, A. K., & Neergaard, J. S. (2025). Development and validation of the Child Hemophilia Treatment Experience Measure: A new observer-reported outcome measure. Haemophilia : the official journal of the World Federation of Hemophilia, 31(1), 48–62. https://doi.org/10.1111/hae.15102
- Mokhtar, F. M., Rajakumar, S., & Zaman Huri, H. (2023). Adherence tool for prophylactic haemophilia treatment in adult and adolescent patients: A systematic review and meta-analysis protocol. PloS one, 18(12), e0289815. https://doi.org/10.1371/journal.pone.0289815
- Wheeler, A. P., Cibelli, E., Hanson, G., Percier, C., Porstmann, T., Shridhar, N., & Shapiro, A. (2025). Treatment and Disease Burden in a Cohort of People With Haemophilia Without Inhibitors in the United States. Haemophilia : the official journal of the World Federation of Hemophilia, 31(5), 912–921. https://doi.org/10.1111/hae.70078
- Hermans, C., & Pierce, G. F. (2025). Therapeutic innovations in hemophilia: the essential role of a positive reinvestment cycle. Blood advances, 9(18), 4631–4639. https://doi.org/10.1182/bloodadvances.2025016497
- Kuijlaars, I. A. R., van der Net, J., Feldman, B. M., Aspdahl, M., Bladen, M., de Boer, W., Cuesta-Barriuso, R., Matlary, R. E. D., Funk, S. M., Hilliard, P., John, J. A., Kempton, C. L., de Kleijn, P., Manco-Johnson, M., Petrini, P., Poonnoose, P., St-Louis, J., Thomas, S., Timmer, M. A., Trakymiene, S. S., … Fischer, K. (2020). Evaluating international Haemophilia Joint Health Score (HJHS) results combined with expert opinion: Options for a shorter HJHS. Haemophilia : the official journal of the World Federation of Hemophilia, 26(6), 1072–1080. https://doi.org/10.1111/hae.14180
- Seoane-Martín, M. E., Cuesta-Barriuso, R., & Rodríguez-Martínez, M. C. (2024). Performance of instrumental activities of daily living in patients with haemophilic arthropathy. A cross-sectional cohort study. Haemophilia : the official journal of the World Federation of Hemophilia, 30(6), 1406–1413. https://doi.org/10.1111/hae.15114
- Saba HI, Roberts HR. Hemostasis and Thrombosis: Practical Guidelines in Clinical Management (Wiley Blackwell). 2014.
- DeLoughery TG. Hemostasis and Thrombosis 4th Edition (Springer). 2019.
- Keohane EM, Otto CN, Walenga JM. Rodak’s Hematology 6th Edition (Saunders). 2019.
- Tiede A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, Di Minno G, d'Oiron R, Salaj P, Jiménez-Yuste V, Huth-Kühne A, Giangrande P (2020). International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica; 105(7). https://doi.org/10.3324/haematol.2019.230771.
- Leebeek FWG, Miesbach W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood (2021) 138 (11): 923–931.
- Berntorp, E., Fischer, K., Hart, D.P. et al. Haemophilia. Nat Rev Dis Primers 7, 45 (2021). https://doi.org/10.1038/s41572-021-00278-x
- Sarmiento Doncel S, Díaz Mosquera GA, Cortes JM, Agudelo Rico C, Meza Cadavid FJ, Peláez RG. Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematol Rep. 2023 Feb 16;15(1):130-150. doi: 10.3390/hematolrep15010014. PMID: 36810557; PMCID: PMC9944491.
- Mehta P, Reddivari AKR. Hemophilia. [Updated 2023 Jun 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551607/
- Young G, Lassila R, Mason J, Prasca S. Deconstructing the ISTH hemophilia guidelines for the clinician. J Thromb Haemost. 2025 May;23(5):1483-1495. doi: 10.1016/j.jtha.2024.11.015. Epub 2024 Nov 29. PMID: 39617189.
- Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, Tiede A, Kessler CM. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017 Jul;92(7):695-705. doi: 10.1002/ajh.24777. Epub 2017 Jun 5. PMID: 28470674.
- Batorova A, Boban A, Brinza M, Lissitchkov T, Nemes L, Zupan Preložnik I, Smejkal P, Zozulya N, Windyga J. Expert opinion on current and future prophylaxis therapies aimed at improving protection for people with hemophilia A. J Med Life. 2022 Apr;15(4):570-578. doi: 10.25122/jml-2022-0103. PMID: 35646171; PMCID: PMC9126455.