Key Takeaways
Hemophilia is an inherited disorder of secondary hemostasis caused by a deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B); both result in delayed, deep tissue bleeding rather than immediate surface bleeding.
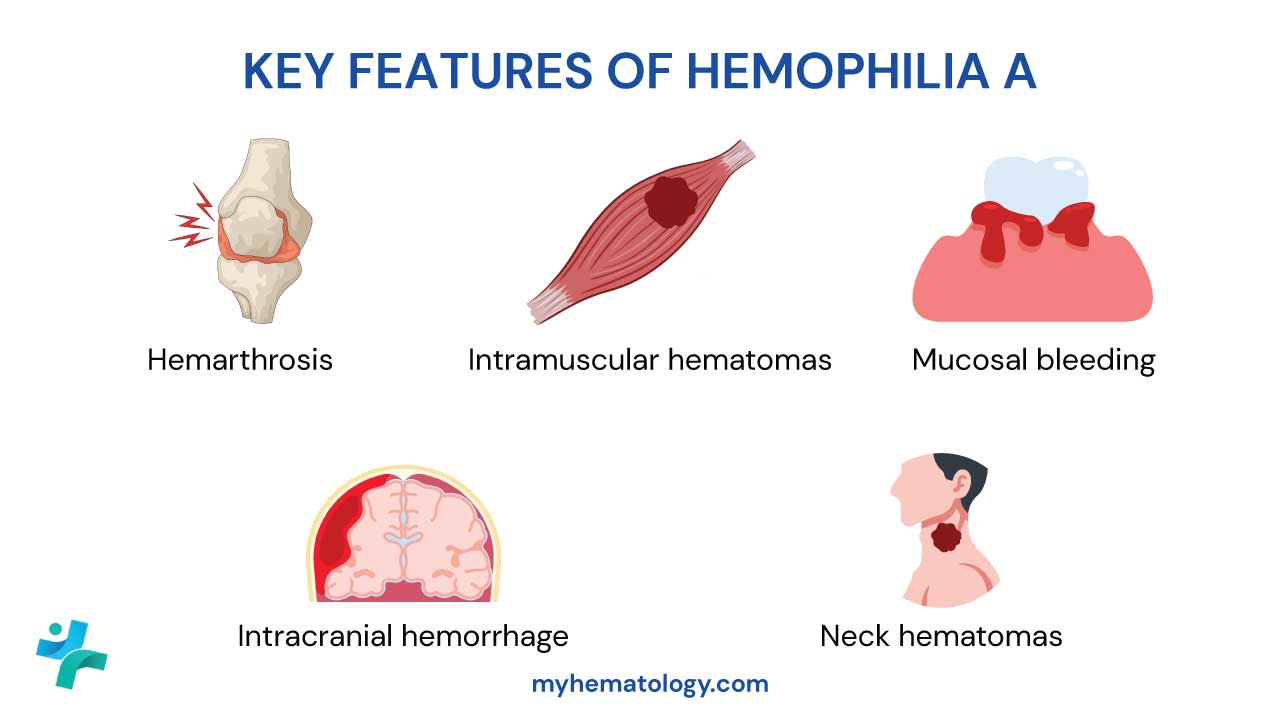
- Clinical features ▾: Joint and soft tissue bleeds as well as excessive bruising. Recurrent painful hemarthrosis and muscle hematomas may lead to progressive joint deformity and disability if poorly treated.
- Laboratory diagnosis ▾: The classic laboratory pattern is a prolonged APTT with a normal PT, normal platelet count, and normal bleeding time; a specific factor assay confirms the type and severity. Severe hemophilia is defined by less than 1% factor activity, moderate by 1% to 5%, and mild by more than 5% to less than 40% [3].
- Treatment and Management ▾: Modern treatment includes recombinant factor concentrates, extended half-life products, emicizumab (for hemophilia A, including with inhibitors), the newer agents fitusiran and marstacimab, and approved gene therapies for both hemophilia A (valoctocogene roxaparvovec) and hemophilia B (etranacogene dezaparvovec) [4,8,9,10].
*Click ▾ for more information
What is hemophilia?
Hemophilia is an inherited bleeding disorder where the blood cannot form a stable clot because a specific clotting protein is missing or faulty. The result is prolonged bleeding, often deep inside joints and muscles, and sometimes without any obvious injury [1].
The body normally stops bleeding in two stages. First, platelets clump at the site of damage to form a temporary plug. Then a chain of clotting proteins reinforces that plug with a tough fibrin mesh. A process known as secondary hemostasis. In hemophilia, the first stage works fine. The second stage breaks down. The plug forms but cannot be locked in place, so bleeding restarts and continues.
Two clotting proteins account for almost all cases. Hemophilia A is caused by a shortage of factor VIII. Hemophilia B, also called Christmas disease, is caused by a shortage of factor IX. Hemophilia A is about four times more common than hemophilia B [1]. Worldwide, the World Federation of Hemophilia estimates roughly 17.1 cases of hemophilia A and 3.8 cases of hemophilia B per 100,000 males, though the true number is higher in countries with stronger diagnostic systems [11].
This article covers the genetics, mechanism, diagnosis, and modern treatment of hemophilia A and B, with a brief comparison to the much rarer hemophilia C at the end.
Classification of Hemophilia
Hemophilia is classified two ways: by the missing factor, and by how much of that factor is still active in the blood.
| Classification | Missing Factor | Other Name |
|---|---|---|
| Hemophilia A (Classic Hemophilia) | Factor VIII (FVIII) | Classic hemophilia |
| Hemophilia B (Christmas Disease) | Factor IX (FIX) | Christmas disease |
Both types follow the same pattern of inheritance and produce indistinguishable symptoms. They can only be told apart with a specific factor assay [7].
Severity is graded by the percentage of normal factor activity present in plasma. This grading shapes how patients bleed and how they are treated [3].
| Severity Level | Factor Activity Level | Typical Bleeding |
|---|---|---|
| Severe | < 1% of normal activity | Spontaneous bleeds, often starting in infancy |
| Moderate | 1%–5% of normal activity | Bleeds after minor injuries or procedures |
| Mild | > 5%–40% of normal activity | Bleeds only after significant trauma or surgery |
Causes and Inheritance of Hemophilia
How hemophilia is passed down
Hemophilia A and B are X-linked recessive disorders, which means the faulty gene sits on the X chromosome. This single fact explains almost all of the inheritance pattern.
Males have one X and one Y chromosome. If their single X carries the faulty gene, there is no backup copy. They have hemophilia.
Females have two X chromosomes. If only one carries the faulty gene, the second X usually produces enough factor to prevent serious bleeding. These women are called carriers. Each of their sons has a 50% chance of inheriting the disorder, and each daughter has a 50% chance of being a carrier.
It is a mistake to assume all carriers are symptom-free. Because of a process called lyonization (random X-inactivation), one X chromosome is switched off in each cell. If most cells happen to silence the working X, factor levels drop. Many carriers have factor activity in the 40% to 60% range, but some run lower and bleed with periods, surgery, or childbirth. The current preferred terminology is "women and girls with hemophilia" rather than "carriers" alone [1].
A female with the faulty gene on both X chromosomes will have full hemophilia, but this is rare and usually involves specific chromosomal arrangements.
About one in three new diagnoses occurs in families with no prior history. These cases are due to spontaneous mutations in the egg, sperm, or developing embryo [7].

Gene Location
The location of the mutation determines the type.
Hemophilia A is caused by mutations in the F8 gene at Xq28; the most common severe mutation is an inversion that disrupts intron 22.
Hemophilia B is caused by mutations in the F9 gene at Xq27, usually point mutations or small deletions [1].
Genetic testing now plays a routine role in carrier identification, prenatal diagnosis, and reproductive planning. Options for at-risk couples include preimplantation genetic testing during IVF and, in pregnancy, chorionic villus sampling or non-invasive prenatal testing.
Acquired Hemophilia
Not every case is inherited. Acquired hemophilia is a rare autoimmune disorder, usually in adults over 60, where the immune system suddenly produces antibodies against the patient's own factor VIII [5]. There is no family history. Bleeding tends to be severe and life-threatening, and standard factor replacement may not work because the antibodies neutralize it. Acquired hemophilia is often linked to underlying cancer, autoimmune disease, or recent pregnancy [6].
Acquired Hemophilia Signs
An older adult with sudden, severe bleeding and no bleeding history. It needs urgent specialist care.
Pathophysiology
The deeper mechanism behind hemophilia is a failure to generate enough thrombin, the enzyme that turns soluble fibrinogen into the fibrin mesh of a stable clot.
The two phases of hemostasis
Primary hemostasis is the first response to a cut: blood vessels narrow, and platelets gather to form a soft plug. Factors VIII and IX play no role here, so this phase is normal in hemophilia. Secondary hemostasis is the chemical chain reaction that follows. Clotting factors activate one another in sequence and reinforce the platelet plug with cross-linked fibrin. This is where hemophilia hits.
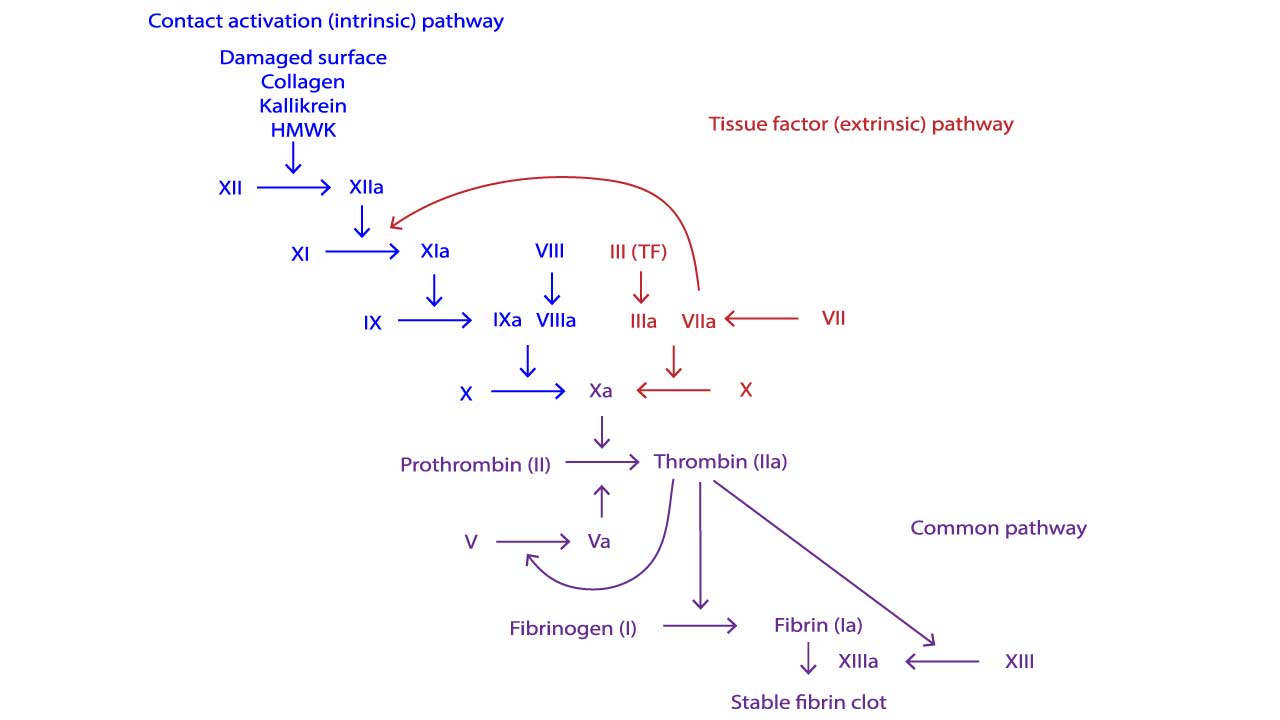
The tenase complex: the bottleneck
The intrinsic pathway of the clotting cascade depends on a partnership between activated factor IX (the enzyme) and activated factor VIII (its helper protein). Together on the surface of a platelet, they form the tenase complex, which is the engine that activates factor X. Factor Xa then drives the rest of the cascade and produces thrombin.
Take away factor VIII and factor IXa loses its helper. Take away factor IX and there is no enzyme to activate. Either way, the tenase complex cannot do its job, factor X is barely activated, thrombin generation collapses, and the fibrin mesh fails to set [1]. Factor VIIIa rapidly activates Factor X to Factor Xa. Factor Xa is the starting point of the common pathway.
Why bleeds happen where they do
This mechanism explains the signature pattern of hemophilia bleeding. Cuts and small injuries seem to stop, then start again hours later as the unstable plug gives way. Bleeding settles into deep, high-pressure spaces where platelet plugs alone cannot hold:
- Joints (hemarthrosis), most often knees, elbows, and ankles.
- Large muscle groups, particularly the iliopsoas and thigh muscles.
- Inside the skull (intracranial hemorrhage), the most dangerous of all.
The contrast with platelet disorders is useful. Platelet problems cause immediate bleeding from skin and mucous membranes (nosebleeds, gum bleeding, easy bruising). Hemophilia causes delayed, deep bleeds.
Signs and Symptoms
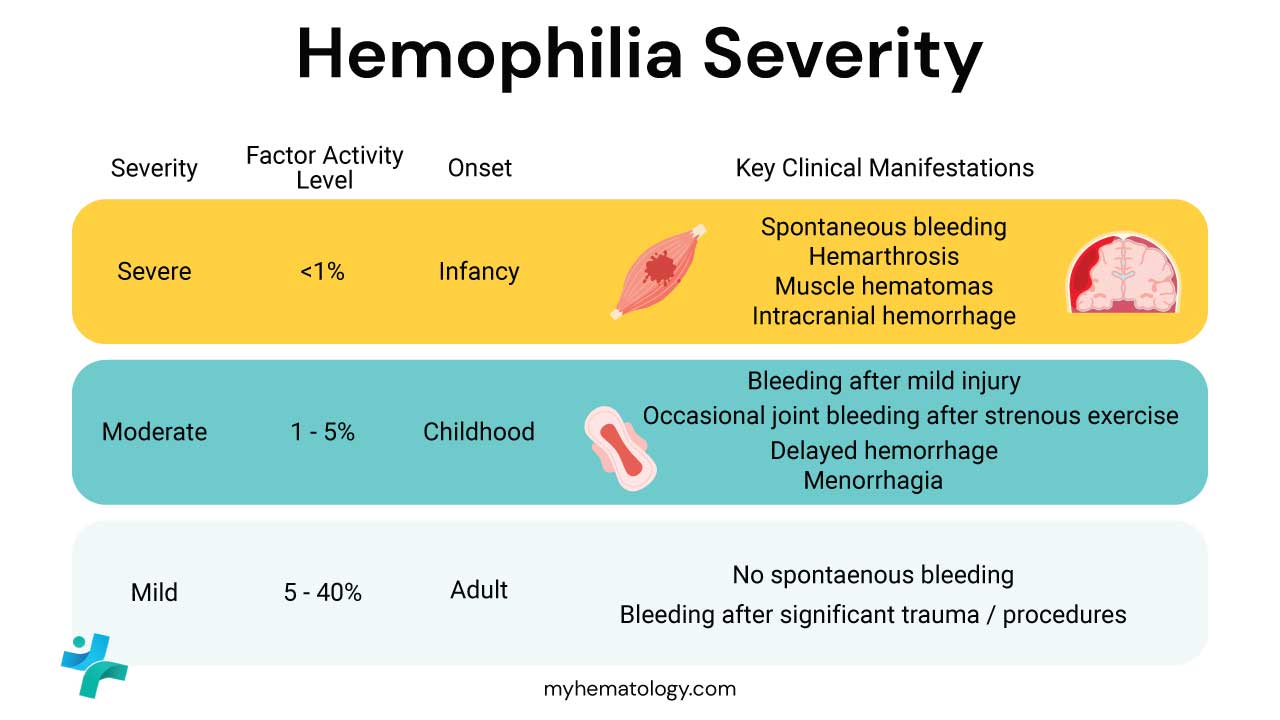
The clinical picture depends almost entirely on residual factor activity.
- Severe hemophilia (less than 1%) produces spontaneous bleeds starting in infancy.
- Moderate hemophilia (1% to 5%) bleeds mostly after minor trauma or procedures.
- Mild hemophilia (more than 5% to less than 40%) may go undetected until adulthood, surfacing only after surgery, dental work, or major injury [3,7].
It is increasingly recognized, however, that a patient's clinical phenotype may not perfectly match their laboratory phenotype. Some patients with 2% factor activity bleed spontaneously like those with severe disease, while some with 0.5% rarely bleed. Modern management increasingly tailors treatment to the individual's bleeding history rather than relying strictly on the laboratory grade [12].
Musculoskeletal Bleeding (The Primary Concern)
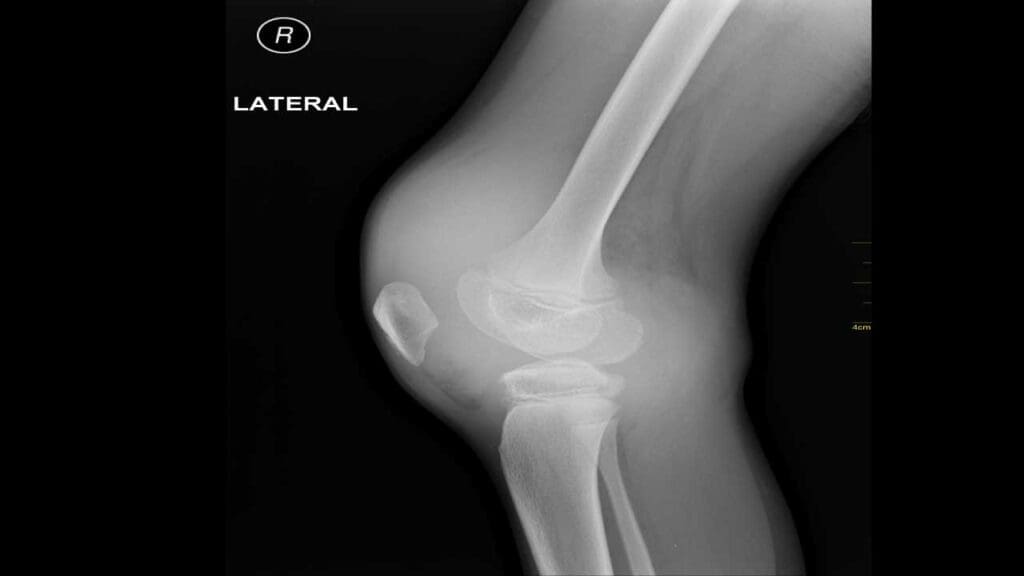
Hemarthrosis is the signature symptom. Blood enters a joint capsule and causes pain, swelling, warmth, and reduced movement. Repeated bleeds into the same joint create a "target joint" and trigger chronic inflammation, cartilage loss, and bone erosion. Over years, this causes hemophilic arthropathy, a destructive joint disease that drives much of the long-term disability in hemophilia [1].
Deep muscle hematomas are the other major concern. Bleeding into a large muscle, such as the iliopsoas, can compress nearby nerves. An iliopsoas bleed compressing the femoral nerve, for example, causes severe groin pain and weakness in the leg.
Surface bleeds
These are less specific to hemophilia but common: nosebleeds (epistaxis), bleeding from the gums during dental work, and large tender bruises (ecchymoses) after minor bumps.
Bleeding emergencies
Some bleeds need immediate factor replacement and emergency assessment:
- Intracranial hemorrhage (ICH) is the leading cause of death in hemophilia. Head injury or unexplained severe headache, vomiting, drowsiness, or seizures must be treated as ICH until a scan rules it out.
- Retroperitoneal bleeding behind the abdominal lining can hide large blood loss.
- Neck or throat bleeds can swell enough to block the airway.
First signs in babies
In severe hemophilia, the disorder usually announces itself within the first two years. Common presenting signs include prolonged bleeding after circumcision, a swelling under the scalp after delivery (cephalohematoma), and unusual bruising as the baby starts to crawl or walk.
Laboratory Investigations and Diagnosis
Diagnosing hemophilia combines clinical suspicion with a recognizable pattern of coagulation tests, followed by a specific factor assay.
Screening tests
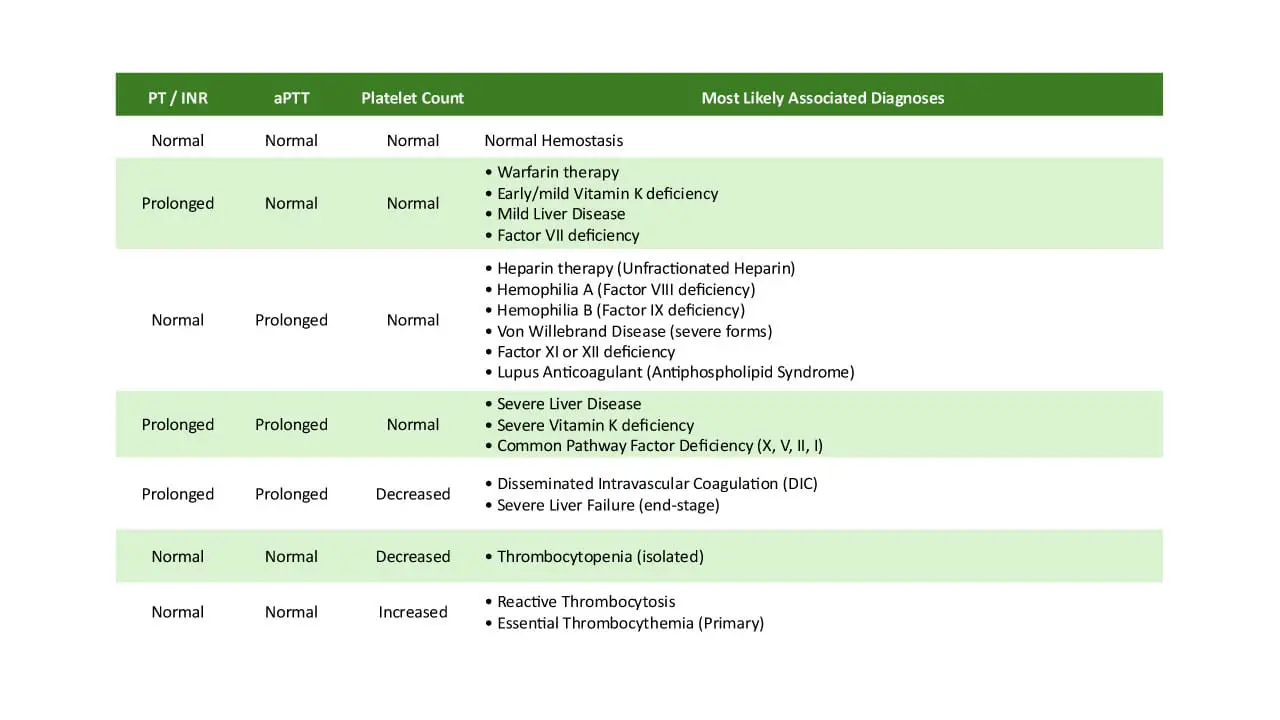
The hallmark coagulation pattern is a prolonged APTT with a normal PT. Here is why.
- Activated partial thromboplastin time (APTT): prolonged. APTT measures the intrinsic and common pathways. Factors VIII and IX both sit in the intrinsic pathway, so their deficiency slows this test down. The longer the APTT, the more severe the deficiency tends to be.
- Prothrombin time (PT): normal. PT measures the extrinsic and common pathways, which do not depend on factor VIII or IX.
- Thrombin time, platelet count, and bleeding time: normal. Hemophilia does not affect platelets, fibrinogen, or thrombin itself.
This combination ─ prolonged APTT, normal PT, normal platelets ─ is the screening signature that triggers further testing.
Confirmatory tests
A specific factor assay measures the activity of factor VIII and factor IX directly. A low factor VIII level confirms hemophilia A; a low factor IX level confirms hemophilia B. The percentage of normal activity sets the severity grade.
Genetic analysis of the F8 and F9 genes is used for carrier screening, prenatal diagnosis, and assessing inhibitor risk, particularly for the intron 22 inversion in F8 [1].
The advent of novel non-factor therapies has introduced new complexities to laboratory testing. Emicizumab artificially shortens the APTT, causing standard one-stage APTT-based factor VIII assays to erroneously read as normal or highly elevated. To accurately measure native factor VIII or detect inhibitors in patients on emicizumab, laboratories must use bovine chromogenic assays, which do not react with the drug [13]. Furthermore, as therapies like fitusiran and anti-TFPI monoclonal antibodies rebalance the coagulation cascade rather than replacing a specific factor, specialized treatment centers increasingly rely on global coagulation assays, such as thrombin generation assays (TGAs) and thromboelastography, to monitor overall clotting capacity [14].
Watching for inhibitors
The most serious complication of treatment is the development of an inhibitor, an antibody that neutralizes infused factor concentrate. Inhibitors are detected and quantified by the Bethesda assay (or the Nijmegen-Bethesda variant). One Bethesda Unit is the amount of inhibitor that wipes out half the factor activity in a standard plasma sample after two hours of incubation. Once inhibitors appear, standard factor replacement stops working and the treatment plan changes [7].
Inhibitors are roughly six to ten times more common in severe hemophilia A (around 20% to 30%) than in severe hemophilia B (around 3% to 5%) [7].
Summary of Expected Laboratory Results
| Test | Result in hemophilia | Rationale |
|---|---|---|
| aPTT | Prolonged | Intrinsic pathway depends on FVIII and FIX |
| PT/TT | Normal | Extrinsic pathway is intact |
| Thrombin time | Normal | Fibrinogen and thrombin are unaffected |
| Platelet Count | Normal | Primary hemostasis is intact |
| Factor VIII Assay | Low in Hemophilia A | Confirms the type and severity |
| Factor IX Assay | Low in Hemophilia B | Confirms the type and severity |
Treatment and Management
The aim of hemophilia care is to prevent bleeds wherever possible and treat them quickly when they happen, so that joints stay healthy and life-threatening bleeds are caught early. Most of this care is delivered through specialized comprehensive hemophilia treatment centers (HTCs), which coordinate hematology, physiotherapy, dentistry, social work, and psychology in one place.
Factor replacement: the foundation
The core treatment for both types is replacing the missing factor.
Recombinant factors are made in genetically engineered cell lines and carry essentially no risk of transmitting blood-borne infection. They are now the preferred choice in countries that can afford them.
Plasma-derived factors, purified from pooled donor plasma, are still used and have an excellent modern safety record after viral inactivation steps. The shift away from plasma-derived products was driven by the contamination tragedies of the 1980s, when many people with hemophilia acquired HIV and hepatitis C from infected concentrates.
Factor concentrates come in two formats:
- Standard half-life (SHL) products require frequent infusions, typically three times a week for factor VIII and twice a week for factor IX.
- Extended half-life (EHL) products are modified to stay active longer in the bloodstream, often allowing weekly or even less frequent dosing. The half-life extension is generally larger for factor IX than for factor VIII.
Prophylaxis ─ regular preventive infusions ─ is the standard of care for severe hemophilia, ideally started in early childhood before joint damage begins. Older guidelines aimed only to keep trough factor levels above 1%. Current ISTH guidance favors higher targets, often 3% to 5% trough or above, with some centers individualizing toward levels that keep the patient bleed-free during normal activity [3].
On-demand treatment ─ infusing factor only when a bleed occurs ─ is reserved for milder disease or for patients who cannot access prophylaxis.
Beyond factor replacement
Several treatments do not involve infusing the missing factor itself.
Desmopressin (DDAVP) is a synthetic version of vasopressin. It triggers the release of stored factor VIII and von Willebrand factor from blood vessel walls. DDAVP is a first-line option for many people with mild hemophilia A. It does nothing for hemophilia B and is not enough for severe hemophilia A [7].
Antifibrinolytics such as tranexamic acid and epsilon-aminocaproic acid stop a clot from breaking down too soon. They are useful for bleeds in the mouth or nose, often added during dental procedures, but should never be used alone for joint or muscle bleeds.
Emicizumab (Hemlibra) is a bispecific antibody that physically links factor IXa and factor X, mimicking the bridging job of factor VIII. It is given as a subcutaneous injection (under the skin), as infrequently as once a month, and works in patients with or without inhibitors. For people with severe hemophilia A and inhibitors in particular, emicizumab has changed daily life, replacing complex IV regimens with a simple injection and dramatically reducing bleeds [8]. It is approved only for hemophilia A.
Fitusiran (Qfitlia) is a small interfering RNA therapy that silences production of antithrombin, a natural anticoagulant. With less antithrombin around, more thrombin is generated and clotting improves. Fitusiran was approved by the FDA in 2025 for hemophilia A and B, with or without inhibitors.
Anti-TFPI monoclonal antibodies block tissue factor pathway inhibitor (TFPI), a natural brake on clotting, thereby boosting hemostasis. Marstacimab (Hympavzi) and concizumab (Alhemo) are regulatory-approved subcutaneous options for patients with hemophilia, marking a major shift toward "rebalancing" the coagulation cascade [15].
Next-generation bispecific antibodies, such as Mim8, have emerged following the success of emicizumab. Clinical trials demonstrate high efficacy, potent thrombin generation, and highly flexible subcutaneous dosing schedules for patients with hemophilia A [16].
Gene therapy is now a clinical reality, not just a future hope. Two AAV-based therapies have regulatory approval [4,9,10]:
- Valoctocogene roxaparvovec (Roctavian) for severe hemophilia A.
- Etranacogene dezaparvovec (Hemgenix) for hemophilia B.
Both deliver a working copy of the relevant gene to liver cells, which then produce the missing factor on their own. A single infusion can free many patients from regular prophylaxis. Long-term durability, the ability to re-treat, and use in children are still being studied.
Real-world data and extended trial follow-ups have revealed differing long-term outlooks for these two therapies. While etranacogene dezaparvovec for hemophilia B has demonstrated highly stable and durable factor IX expression over several years, factor VIII expression from valoctocogene roxaparvovec for hemophilia A has been shown to gradually wane over 3 to 5 years in a significant portion of patients, requiring some to eventually resume prophylaxis [17].
Pain and joint care
Pain control deserves special attention. NSAIDs are generally avoided in hemophilia because they affect platelet function and can worsen bleeding; paracetamol (acetaminophen) and selective COX-2 inhibitors are usually preferred, with input from the treating team.
For acute joint bleeds, factor (or emicizumab cover) is given immediately, followed by the RICE protocol: rest, ice, compression, and elevation. Once the bleed settles, physical therapy rebuilds range of motion and surrounding muscle strength. For end-stage hemophilic arthropathy, orthopedic procedures such as synovectomy or joint replacement may be needed.
Hemophilia C: Factor XI Deficiency
Hemophilia C is a separate disorder caused by factor XI deficiency. Despite the shared name, it behaves differently from hemophilia A and B in three key ways. Inheritance is autosomal recessive, not X-linked, so it affects males and females equally. Bleeding is usually triggered by surgery or trauma rather than happening spontaneously. And factor XI levels do not predict bleeding well, meaning people with similar lab numbers can have very different clinical courses.
| Feature | Hemophilia A | Hemophilia B | Hemophilia C |
| Missing factor | Factor VIII | Factor IX | Factor XI |
| Inheritance | X-Linked Recessive | X-Linked Recessive | Autosomal Recessive |
| Bleeding Pattern | Spontaneous, deep tissue | Spontaneous, deep tissue | Mostly post-surgical or post-traumatic |
| Standard Prophylaxis | FVIII concentrate or emicizumab | FIX concentrate | Usually not needed |
| Acute Treatment | IV FVIII concentrate | IV FIX concentrate | Tranexamic acid; FXI concentrate or FFP for major bleeds |
| Inhibitor risk | High (~20–30% in severe disease) | Low (~3–5% in severe disease) | Very rare |
Frequently Asked Questions (FAQs)
Can a person with hemophilia live a normal life?
Yes. With access to modern treatment, most people with hemophilia complete their education, work, raise families, and participate in many sports. Prophylactic factor or emicizumab keeps bleeding rates very low, and gene therapy now offers some patients freedom from regular treatment altogether. The biggest factor is access. In countries with strong hemophilia care systems, life expectancy approaches that of the general male population. In low-resource settings, the picture is harder.
Is hemophilia life threatening?
It can be, particularly when untreated. The most dangerous bleeds are those into the brain, the airway, and the abdomen. With prompt factor replacement and modern prophylaxis, the risk of a fatal bleed is much lower than it once was, but never zero. Any head injury in a person with hemophilia should be treated as a potential intracranial bleed until proven otherwise.
Can hemophilia get worse with age?
The severity of the underlying deficiency does not change, but two age-related issues do come into play. First, joint damage from earlier bleeds tends to accumulate, and pain and disability can worsen even with good treatment. Second, some patients develop inhibitors over time, which complicates therapy. Newer agents such as emicizumab, and gene therapy, have shifted this picture for many older patients.
Can hemophiliacs donate blood?
In most blood donation systems, no. Donation policies are designed to protect both donors and recipients. People with hemophilia are advised against donating blood because of bleeding risk at the venipuncture site, and because they often rely on plasma-derived products themselves.
Is hemophilia painful?
It can be very painful, especially during a joint bleed and during chronic flares of hemophilic arthropathy. Pain is one of the leading drivers of reduced quality of life in adults with hemophilia. Pain control is built into modern care, and importantly, NSAIDs like ibuprofen are usually avoided because they worsen bleeding. Paracetamol (acetaminophen) and selective COX-2 inhibitors are generally preferred.
Why is the APTT prolonged in hemophilia while the PT is normal?
The PT measures the extrinsic and common pathways, which depend on factor VII and tissue factor and are not affected by hemophilia. The APTT measures the intrinsic and common pathways, which run through factors VIII and IX. When either factor is low, the intrinsic pathway slows down and the APTT comes back prolonged. This pattern, with normal platelets and normal PT, is the classic screening signature that prompts a specific factor assay.
Can women have hemophilia?
Yes. Women who carry one faulty copy of the gene can have low factor levels because of lyonization, the random switching off of one X chromosome in each cell. Symptoms include heavy menstrual bleeding, postpartum bleeding, and excessive bleeding after surgery. Rarely, a woman inherits faulty genes on both X chromosomes and has full hemophilia. The terminology has shifted from "carrier" alone to "women and girls with hemophilia" when factor levels are low enough to cause bleeding [1].
When is emicizumab the right choice for hemophilia A?
Emicizumab is particularly suited to patients with severe hemophilia A who have inhibitors, who struggle with the burden of frequent IV infusions, who have poor venous access, or who want less frequent dosing. It dramatically reduces bleeding rates and is given as a subcutaneous injection as infrequently as once a month [8]. It does not work for hemophilia B.
What is the rationale behind extended half-life factors?
The goal is fewer needles. Modifications such as Fc fusion or PEGylation slow how quickly the factor is cleared from the body. EHL products are available for both factor VIII and factor IX. The half-life extension is more pronounced for factor IX, where weekly or longer dosing intervals are achievable, than for factor VIII.
Is there a cure for hemophilia?
Yes, in a meaningful sense. Two AAV-based gene therapies are now approved: valoctocogene roxaparvovec (Roctavian) for hemophilia A and etranacogene dezaparvovec (Hemgenix) for hemophilia B. A single infusion can raise factor levels enough that many treated patients no longer need regular prophylaxis. Long-term durability is still being studied, and gene therapy is not suitable for everyone, including children and people with certain pre-existing antibodies to the viral vector. Prophylaxis with factor concentrate or emicizumab remains the standard for most patients.
What activities should someone with hemophilia avoid?
High-impact contact sports such as boxing, rugby, American football, and ice hockey are generally discouraged because of the head and joint injury risk. Lower-impact activities like swimming, cycling, walking, and many forms of strength training are encouraged and protect joint health. The decision is individual and should be guided by a hemophilia treatment center team, taking into account severity, prophylaxis regimen, and target joints. Activity is important; over-restriction leads to muscle weakness and worse joint outcomes.
Glossary of Medical Terms
- Hemarthrosis — bleeding inside a joint, the defining feature of severe hemophilia.
- Prophylaxis — scheduled preventive infusions or injections to stop bleeds before they start.
- Inhibitor — an antibody the immune system makes against infused clotting factor, which blocks the treatment from working.
- Bethesda Unit (BU) — the unit used to measure inhibitor concentration in plasma.
- Tenase complex — the partnership of activated factor IX (the enzyme) and activated factor VIII (its helper) that activates factor X.
- Emicizumab — an antibody-based therapy that bridges factors IX and X, doing the job of missing factor VIII.
- Hemophilic arthropathy — long-term joint damage caused by repeated joint bleeds.
- Immune tolerance induction (ITI) — high-dose factor therapy aimed at training the immune system to stop producing inhibitors.
- Extended half-life (EHL) factor — a modified factor product that stays active longer, reducing how often infusions are needed.
- Lyonization — random inactivation of one X chromosome in each cell of a female; explains why some carriers have low factor levels and bleed.
- Acquired hemophilia — a rare autoimmune disorder where the body suddenly makes antibodies against its own factor VIII.
- Comprehensive hemophilia treatment center (HTC) — a multidisciplinary clinic that coordinates all aspects of hemophilia care.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Berntorp, E., Fischer, K., Hart, D.P. et al. Haemophilia. Nat Rev Dis Primers 7, 45 (2021). https://doi.org/10.1038/s41572-021-00278-x
- Srivastava, A., Santagostino, E., Dougall, A., Kitchen, S., Sutherland, M., Pipe, S. W., Carcao, M., Mahlangu, J., Ragni, M. V., Windyga, J., Llinás, A., Goddard, N. J., Mohan, R., Poonnoose, P. M., Feldman, B. M., Lewis, S. Z., van den Berg, H. M., Pierce, G. F., & WFH Guidelines for the Management of Hemophilia panelists and co-authors (2020). WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia : the official journal of the World Federation of Hemophilia, 26 Suppl 6, 1–158. https://doi.org/10.1111/hae.14046
- Young, G., Lassila, R., Mason, J., & Prasca, S. (2025). Deconstructing the ISTH hemophilia guidelines for the clinician. Journal of thrombosis and haemostasis : JTH, 23(5), 1483–1495. https://doi.org/10.1016/j.jtha.2024.11.015
- Leebeek, F. W. G., & Miesbach, W. (2021). Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood, 138(11), 923–931. https://doi.org/10.1182/blood.2019003777
- Tiede, A., Collins, P., Knoebl, P., Teitel, J., Kessler, C., Shima, M., Di Minno, G., d'Oiron, R., Salaj, P., Jiménez-Yuste, V., Huth-Kühne, A., & Giangrande, P. (2020). International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica, 105(7), 1791–1801. https://doi.org/10.3324/haematol.2019.230771
- Kruse-Jarres, R., Kempton, C. L., Baudo, F., Collins, P. W., Knoebl, P., Leissinger, C. A., Tiede, A., & Kessler, C. M. (2017). Acquired hemophilia A: Updated review of evidence and treatment guidance. American journal of hematology, 92(7), 695–705. https://doi.org/10.1002/ajh.24777
- Sarmiento Doncel, S., Díaz Mosquera, G. A., Cortes, J. M., Agudelo Rico, C., Meza Cadavid, F. J., & Peláez, R. G. (2023). Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematology reports, 15(1), 130–150. https://doi.org/10.3390/hematolrep15010014
- Mahlangu, J., Oldenburg, J., Paz-Priel, I., Negrier, C., Niggli, M., Mancuso, M. E., Schmitt, C., Jiménez-Yuste, V., Kempton, C., Dhalluin, C., Callaghan, M. U., Bujan, W., Shima, M., Adamkewicz, J. I., Asikanius, E., Levy, G. G., & Kruse-Jarres, R. (2018). Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. The New England journal of medicine, 379(9), 811–822. https://doi.org/10.1056/NEJMoa1803550
- Pipe, S., Leebeek, F., Recht, M., Key, N., Castaman, G., Miesbach, W., Lattimore, S., Peerlinck, K., Van der Valk, P., Coppens, M., Kampmann, P., Meijer, K., O’Connell, N., Pasi, K., Hart, D., Kazmi, R., Astermark, J., Hermans, C., Klamroth, R., … Monahan, P. (2023). Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. In New England Journal Of Medicine (Vol. 388, Issue 8, pp. 706–718). Massachusetts Medical Society. https://doi.org/10.1056/NEJMoa2211644
- Ozelo, M. C., Mahlangu, J., Pasi, K. J., Giermasz, A., Leavitt, A. D., Laffan, M., Symington, E., Quon, D. V., Wang, J. D., Peerlinck, K., Pipe, S. W., Madan, B., Key, N. S., Pierce, G. F., O'Mahony, B., Kaczmarek, R., Henshaw, J., Lawal, A., Jayaram, K., Huang, M., … GENEr8-1 Trial Group (2022). Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. The New England journal of medicine, 386(11), 1013–1025. https://doi.org/10.1056/NEJMoa2113708
- World Federation of Hemophilia. (2024). Report on the Annual Global Survey 2023. World Federation of Hemophilia.
- Blanchette, V. S., Key, N. S., Ljung, L. R., Manco-Johnson, M. J., van den Berg, H. M., Srivastava, A., & Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis (2014). Definitions in hemophilia: communication from the SSC of the ISTH. Journal of thrombosis and haemostasis : JTH, 12(11), 1935–1939. https://doi.org/10.1111/jth.12672
- Müller, J., Pekrul, I., Pötzsch, B., Berning, B., Oldenburg, J., & Spannagl, M. (2019). Laboratory Monitoring in Emicizumab-Treated Persons with Hemophilia A. Thrombosis and haemostasis, 119(9), 1384–1393. https://doi.org/10.1055/s-0039-1692427
- Lenting P. J. (2020). Laboratory monitoring of hemophilia A treatments: new challenges. Blood advances, 4(9), 2111–2118. https://doi.org/10.1182/bloodadvances.2019000849
- Matsushita, T., Shapiro, A., Abraham, A., Angchaisuksiri, P., Castaman, G., Cepo, K., d'Oiron, R., Frei-Jones, M., Goh, A. S., Haaning, J., Hald Jacobsen, S., Mahlangu, J., Mathias, M., Nogami, K., Skovgaard Rasmussen, J., Stasyshyn, O., Tran, H., Vilchevska, K., Villarreal Martinez, L., Windyga, J., … explorer7 Investigators (2023). Phase 3 Trial of Concizumab in Hemophilia with Inhibitors. The New England journal of medicine, 389(9), 783–794. https://doi.org/10.1056/NEJMoa2216455
- Chowdary, P., Lentz, S. R., Gil, L., López-Jaime, F. J., Windyga, J., Ong Clausen, W. H., Laursen, P. N., & Mahlangu, J. (2025). FRONTIER1 multiple ascending dose extension: a safety, tolerability, pharmacokinetics, and pharmacodynamics study of Mim8 in people with hemophilia A. Research and practice in thrombosis and haemostasis, 9(7), 103207. https://doi.org/10.1016/j.rpth.2025.103207
- Madan, B., Ozelo, M. C., Raheja, P., Symington, E., Quon, D. V., Leavitt, A. D., Pipe, S. W., Lowe, G., Kenet, G., Reding, M. T., Mason, J., Wang, M., von Drygalski, A., Klamroth, R., Shapiro, S., Chambost, H., Dunn, A. L., Oldenburg, J., Chou, S. C., Peyvandi, F., … Mahlangu, J. (2024). Three-year outcomes of valoctocogene roxaparvovec gene therapy for hemophilia A. Journal of thrombosis and haemostasis : JTH, 22(7), 1880–1893. https://doi.org/10.1016/j.jtha.2024.04.001