Key Takeaways
Ehlers-Danlos Syndrome (EDS) is a group of genetic connective tissue disorders characterized by:
- Joint hypermobility: Excessive range of motion in joints, leading to pain, instability, and dislocations.
- Skin hyperextensibility: Skin that can be stretched to an abnormal extent, often with easy bruising and delayed wound healing.
- Tissue fragility: Increased risk of tissue damage, including blood vessels, organs, and joints.
Types of EDS ▾
- Hypermobile EDS (hEDS): Most common, characterized by joint hypermobility and skin elasticity.
- Classical EDS (cEDS): Characterized by skin hyperextensibility, joint hypermobility, and fragile tissues.
- Vascular EDS (vEDS): Rare but life-threatening, associated with fragile blood vessels.
- Other subtypes: Several less common types exist.
Diagnosis ▾
- Based on clinical evaluation, genetic testing, and other medical assessments.
Management ▾
- Focuses on managing symptoms, preventing complications, and improving quality of life.
- May involve physical therapy, pain management, surgical interventions, and genetic counseling.
*Click ▾ for more information
Introduction
Ehlers-Danlos Syndrome (EDS) is a group of inherited connective tissue disorders characterized by hyperelasticity of the skin, joint hypermobility, and other connective tissue abnormalities. These disorders are caused by defects in the genes that produce collagen, a protein essential for the structure and function of connective tissues.
Historical Overview
The first documented cases of Ehlers-Danlos Syndrome (EDS) can be traced back to the 18th century. However, it wasn’t until the early 20th century that the condition was more formally recognized and described. The French dermatologist Pierre Ehlers and the American surgeon William Danlos independently identified and described cases of this syndrome, leading to its eponymous name.
In recent decades, advances in genetics have led to the identification of specific genetic mutations associated with different types of Ehlers-Danlos Syndrome (EDS). This has significantly improved our understanding of the underlying causes and mechanisms of this disorder.
Causes and Pathogenesis
Ehlers-Danlos Syndrome (EDS) is primarily caused by genetic mutations that affect the structure or function of collagen or other connective tissue proteins. These proteins play a crucial role in providing structural support to the body’s tissues, including skin, joints, and blood vessels.
Types of Ehlers-Danlos Syndrome (EDS) and Their Associated Genetic Mutations
There are several subtypes of Ehlers-Danlos Syndrome (EDS), each characterized by distinct clinical features and genetic mutations.
- Hypermobile EDS (hEDS): This is the most common type of Ehlers-Danlos Syndrome (EDS), characterized by excessive joint flexibility. While the genetic causes of hEDS are complex and often involve multiple genes, mutations in genes related to collagen synthesis or cross-linking, such as COL5A1 and COL5A2, have been implicated.
- Classical EDS (cEDS): Characterized by skin hyperextensibility, joint hypermobility, and easy bruising. Mutations in the COL3A1 gene, which encodes collagen type III, are the primary cause of cEDS.
- Vascular EDS (vEDS): A rare but potentially life-threatening subtype characterized by thin, fragile blood vessels that can rupture and lead to internal bleeding. Mutations in the COL3A1 gene, which encodes collagen type III, are the primary cause of vEDS.
- Kyphoscoliosis EDS (kEDS): Characterized by excessive curvature of the spine (kyphosis and scoliosis). Genetic mutations in the PLOD2 gene, which encodes lysyl oxidase, are associated with kEDS.
- Arthrochalasia EDS (aEDS): Characterized by joint hypermobility and contractures. Genetic mutations in the TNXB gene, which encodes tenascin-X, are associated with aEDS.
- Other subtypes: Additional subtypes, such as dermatosparaxis EDS and myopathic EDS, are characterized by specific clinical features and genetic mutations.
Pathophysiological Mechanisms of EDS
The genetic mutations associated with Ehlers-Danlos Syndrome (EDS) lead to defects in the structure or function of collagen or other connective tissue proteins. Defects in the cross-linking of collagen fibers can impair the structural integrity of connective tissue. Reduced tensile strength and elasticity of connective tissue can lead to joint hypermobility, skin hyperextensibility, and easy bruising.
This can result in:
- Vascular abnormalities: In vEDS, mutations in COL3A1 can lead to the formation of thin, fragile blood vessels that are prone to rupture.
- Skeletal abnormalities: In kEDS and aEDS, mutations in PLOD2 and TNXB, respectively, can affect the development and structure of the skeleton.
- Joint instability: The hypermobility seen in many types of EDS is due to weakened joint ligaments and capsules.
- Skin fragility: Mutations in collagen genes can also lead to thin, fragile skin that is prone to bruising and tearing.
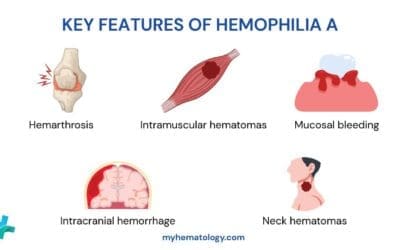
Ehlers-Danlos Syndrome (EDS) Symptoms
The Ehlers-Danlos Syndrome (EDS) symptoms can vary significantly depending on the specific subtype. However, there are some common EDS symptoms that are often observed across different types.
Joint Hypermobility
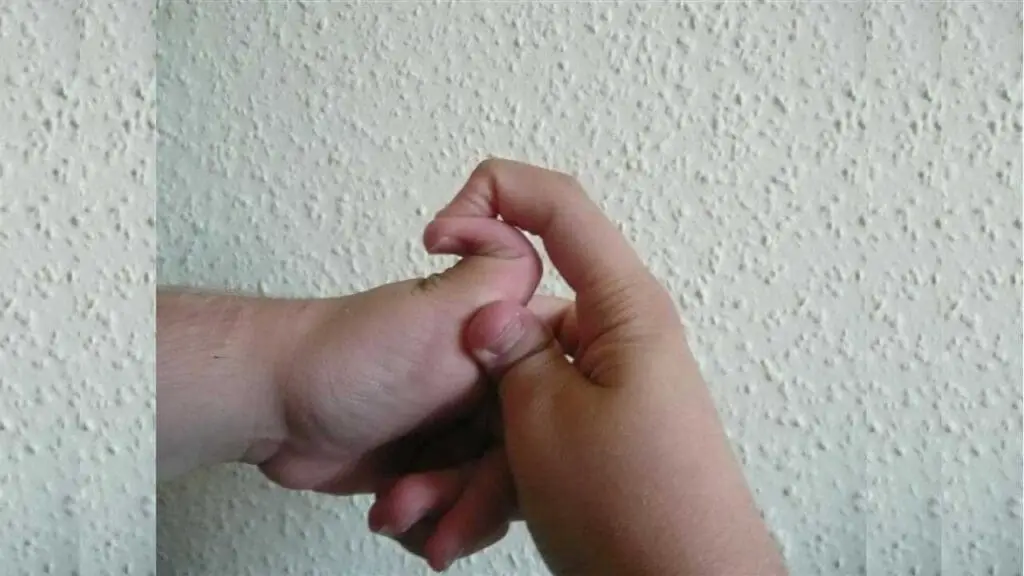
- Excessive range of motion: Individuals with Ehlers-Danlos Syndrome (EDS) often have abnormally flexible joints, allowing for greater than normal movement.
- Joint pain and instability: The hypermobility can lead to joint pain, instability, and dislocation.
- Fatigue: The constant strain on joints can contribute to fatigue and decreased physical endurance.
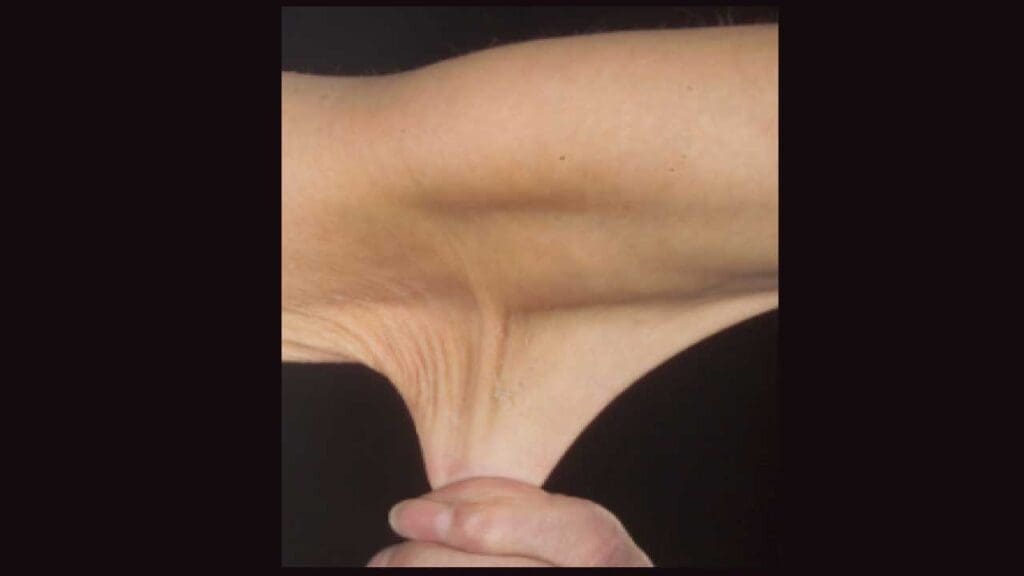
Skin Hyperextensibility
- Excessive elasticity: The skin of individuals withEhlers-Danlos Syndrome (EDS) is often unusually elastic, allowing it to be stretched to a greater extent than normal.
- Easy bruising: The thin, fragile skin can lead to easy bruising and bleeding.
- Delayed wound healing: Wounds may take longer to heal due to impaired tissue repair.
Other Potential EDS Symptoms
- Gastrointestinal issues: Individuals with Ehlers-Danlos Syndrome (EDS) may experience gastrointestinal problems, such as constipation, diarrhea, and abdominal pain.
- Cardiovascular complications: Certain types of Ehlers-Danlos Syndrome (EDS), such as vascular EDS, are associated with an increased risk of cardiovascular problems, including aortic aneurysm and rupture.
- Neurological complications: In some cases, Ehlers-Danlos Syndrome (EDS) can be associated with neurological problems, such as nerve pain and spinal instability.
- Psychological impact: The physical limitations and pain associated with Ehlers-Danlos Syndrome (EDS) can have a significant psychological impact, leading to anxiety, depression, and low self-esteem.
EDS Diagnosis
The diagnosis of EDS often involves a combination of clinical evaluation, family history, and specific investigations. The choice of investigations will depend on the suspected subtype and the patient’s clinical presentation.
Clinical Evaluation
- Beighton score: A scoring system used to assess joint hypermobility. A score of 4 or more out of 5 indicates significant joint hypermobility.
- Family history: A thorough family history can help identify other family members with EDS or related conditions.
Laboratory Investigations
- Blood tests: Blood tests like CBC to rule out anemia or other blood disorders, C-reactive protein (CRP) and ESR to indicate inflammation if they are elevated, thyroid function tests to rule out thyroid disorders which can cause joint paint and fatigues and autoimmune markers such as rheumatoid factor or antinuclear antibodies, may be performed.
- Genetic testing: Genetic testing can be used to identify specific mutations associated with different subtypes of Ehlers-Danlos Syndrome (EDS). The choice of genetic testing will depend on the suspected subtype.
- Skin biopsy: A skin biopsy may be performed to assess the structure and composition of the skin. This can be helpful in diagnosing certain subtypes of Ehlers-Danlos Syndrome (EDS).
Imaging Studies
- X-rays: X-rays can be used to assess joint morphology and look for signs of joint instability or arthritis.
- Echocardiogram: An echocardiogram may be performed to evaluate the structure and function of the heart, particularly in patients with suspected vascular EDS.
- MRI: Magnetic resonance imaging (MRI) can provide detailed images of joints, muscles, and other tissues. It can be helpful in assessing joint damage and identifying potential complications.
Other Investigations
- Gastrointestinal endoscopy: If gastrointestinal symptoms are present, an endoscopy may be performed to evaluate the upper or lower gastrointestinal tract.
- Neurological evaluation: A neurological evaluation may be performed to assess for any neurological complications associated with Ehlers-Danlos Syndrome (EDS).
Differential Diagnosis of Ehlers-Danlos Syndrome (EDS)
When evaluating a patient with symptoms suggestive of EDS, it is important to consider other conditions that may present similarly.
- Hypermobility spectrum disorder (HSD): This condition is characterized by joint hypermobility without the other features typically associated with EDS, such as skin hyperextensibility.
- Osteogenesis imperfecta: This is a genetic disorder characterized by fragile bones that are prone to fractures. While it can also present with joint hypermobility, it is typically associated with other features, such as bone deformities and blue sclera.
- Marfan syndrome: This is a genetic disorder characterized by tall stature, long limbs, and cardiovascular abnormalities. It can also present with joint hypermobility.
- Arthrogryposis multiplex congenita (AMC): This is a condition characterized by joint contractures that are present at birth. It can be associated with joint hypermobility in some cases.
- Rheumatoid arthritis: This is an autoimmune disease characterized by joint inflammation and pain. While it can cause joint hypermobility in some cases, it is typically associated with other features, such as morning stiffness and joint swelling.
- Juvenile idiopathic arthritis (JIA): This is a type of arthritis that affects children and adolescents. It can cause joint pain, swelling, and limited range of motion.
EDS Treatment and Management
Ehlers-Danlos Syndrome (EDS) treatment and management focus on managing symptoms, preventing complications, and improving quality of life. The specific approach will vary depending on the subtype of Ehlers-Danlos Syndrome (EDS) and the severity of the patient’s symptoms.
Supportive Care
- Physical therapy and occupational therapy: Physical therapy and occupational therapy can help individuals with Ehlers-Danlos Syndrome (EDS) improve joint function, strength, and flexibility. These therapies can also teach coping strategies for pain management and daily living activities.
- Pain management: Non-pharmacological approaches, such as physical therapy, relaxation techniques, and cognitive-behavioral therapy, can be effective in managing pain. Over-the-counter pain relievers may also be used. In some cases, stronger pain medications may be necessary for EDS treatment.
- Joint protection: Individuals with Ehlers-Danlos Syndrome (EDS) should be encouraged to protect their joints by avoiding activities that can cause excessive stress or strain. Assistive devices, such as braces or splints, may be helpful in providing support to joints.
Surgical Interventions
In some cases, surgical interventions may be necessary to address specific complications of Ehlers-Danlos Syndrome (EDS), such as:
- Joint replacement: Joint replacement surgery may be considered for individuals with severe joint damage or arthritis.
- Aortic aneurysm repair: Individuals with vascular EDS who develop an aortic aneurysm may require surgical repair.
- Decompression surgery: Decompression surgery may be necessary to relieve pressure on nerves in the spinal canal.
Genetic Counseling
Genetic counseling can provide information about the risk of passing Ehlers-Danlos Syndrome (EDS) on to future generations. It can also help families understand the implications of a diagnosis and make informed decisions about reproductive planning.
Lifestyle Modifications
- Weight management: Maintaining a healthy weight can help reduce stress on joints and improve overall health.
- Exercise: Regular exercise, such as swimming or yoga, can help improve strength, flexibility, and balance.
- Stress management: Stress management techniques, such as relaxation exercises and meditation, can help reduce pain and improve overall well-being.
Prognosis
The prognosis of Ehlers-Danlos Syndrome (EDS) varies significantly depending on the specific subtype and the severity of the patient’s symptoms. While there is no cure for Ehlers-Danlos Syndrome (EDS), with appropriate management, many individuals with Ehlers-Danlos Syndrome (EDS) can lead fulfilling lives.
Factors Affecting Prognosis
- Subtype: Some subtypes of Ehlers-Danlos Syndrome (EDS), such as vascular EDS, carry a higher risk of serious complications, including aortic aneurysm and rupture.
- Severity of symptoms: The severity of joint hypermobility, skin fragility, and other symptoms can affect the overall quality of life and the risk of complications.
- Management: The effectiveness of treatment and management strategies can significantly influence the prognosis.
- Individual factors: Factors such as age, general health, and psychological well-being can also impact the prognosis.
Potential Long-Term Complications
- Joint damage and arthritis: Over time, joint hypermobility can lead to joint damage and arthritis, which can cause pain, stiffness, and reduced mobility.
- Cardiovascular complications: Individuals with vascular EDS are at increased risk of aortic aneurysm and rupture, which can be life-threatening.
- Gastrointestinal issues: Chronic gastrointestinal problems can impact quality of life and may require ongoing management.
- Psychological impact: The physical limitations and pain associated with EDS can have a significant psychological impact, leading to anxiety, depression, and low self-esteem.
Frequently Asked Questions (FAQs)
What is the life expectancy of Ehlers-Danlos syndrome?
The life expectancy of individuals with Ehlers-Danlos Syndrome (EDS) can vary significantly depending on the specific subtype and the severity of the condition.
- Hypermobile EDS (hEDS): Individuals with hEDS generally have a normal life expectancy.
- Classical EDS (cEDS): While cEDS can lead to complications, the overall life expectancy is typically normal.
- Vascular EDS (vEDS): This subtype is associated with a higher risk of life-threatening complications, such as aortic aneurysm and rupture. Individuals with vEDS may have a reduced life expectancy.
Does EDS get worse with age?
Yes, Ehlers-Danlos Syndrome (EDS) can worsen with age. As individuals with EDS get older, the symptoms and complications associated with the condition may become more severe. This is particularly true for individuals with vascular EDS, which can lead to serious cardiovascular problems.
Factors that can contribute to the worsening of EDS over time include:
- Joint damage: Repeated stress on joints can lead to damage, inflammation, and arthritis.
- Tissue degeneration: Connective tissues can deteriorate over time, leading to increased skin fragility and other problems.
- Cardiovascular complications: Individuals with vascular EDS are at increased risk of aortic aneurysm and rupture, which can become more severe with age.
It’s important to note that while EDS may worsen with age, the rate of progression can vary significantly among individuals. With appropriate management and medical care, it is possible to mitigate the effects of the condition and improve quality of life.
Can EDS be cured?
No, Ehlers-Danlos Syndrome (EDS) cannot be cured. It is a genetic disorder that is caused by defects in connective tissue proteins.
However, while there is no cure for EDS, it can be managed effectively with appropriate treatment and lifestyle modifications. The goal of treatment is to manage symptoms, prevent complications, and improve quality of life.
Does EDS affect breasts?
Yes, Ehlers-Danlos Syndrome (EDS) can affect breasts. Due to the hyperelasticity of connective tissues in individuals with EDS, the skin supporting the breasts may become less firm, leading to sagging or drooping. Additionally, some individuals with EDS may experience joint hypermobility in the shoulder area, which can affect breast support and contribute to sagging. This is particularly noticeable in individuals with hypermobile EDS, which is characterized by excessive joint flexibility and skin elasticity.
In addition to sagging breasts, EDS can also contribute to other breast-related issues, such as:
- Pain and discomfort: The hypermobility of joints can cause pain and discomfort in the chest area, including the breasts.
- Increased risk of injury: The loose connective tissue in EDS can make the breasts more susceptible to injury, such as bruising or tearing.
- Difficulty with breastfeeding: In some cases, EDS can make it difficult to breastfeed due to the loose skin and connective tissues in the chest area.
What is the inheritance pattern of EDS mutations?
hlers-Danlos Syndrome (EDS) is typically inherited in an autosomal dominant pattern. This means that a single copy of the mutated gene from either parent is sufficient to cause the condition.
In autosomal dominant inheritance, affected individuals have a 50% chance of passing the mutated gene to their children. However, it’s important to note that the severity of EDS can vary significantly among individuals, even within the same family.
While most cases of EDS are inherited in an autosomal dominant pattern, there are also some cases that are inherited in an autosomal recessive pattern. This means that both parents must carry the mutated gene in order for their child to be affected.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Miklovic T, Sieg VC. Ehlers-Danlos Syndrome. [Updated 2023 May 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549814/
- Riley B. The Many Facets of Hypermobile Ehlers-Danlos Syndrome. J Am Osteopath Assoc. 2020 Jan 1;120(1):30-32. doi: 10.7556/jaoa.2020.012. PMID: 31904772.
- Ritelli M, Colombi M. Molecular Genetics and Pathogenesis of Ehlers-Danlos Syndrome and Related Connective Tissue Disorders. Genes (Basel). 2020 May 13;11(5):547. doi: 10.3390/genes11050547. PMID: 32414079; PMCID: PMC7288446.
- Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. 2010 Oct;12(10):597-605. doi: 10.1097/GIM.0b013e3181eed412. PMID: 20847697.
- Engelbert RH, Juul-Kristensen B, Pacey V, de Wandele I, Smeenk S, Woinarosky N, Sabo S, Scheper MC, Russek L, Simmonds JV. The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):158-167. doi: 10.1002/ajmg.c.31545. PMID: 28306230.