Key Takeaways
Hemophilia B is an inherited bleeding disorder caused by missing or faulty Factor IX (FIX), a clotting protein. It is sometimes called Christmas disease, after the first patient described with it.
- Epidemiology ▾: It is the second most common type of hemophilia, making up roughly 15% to 20% of hemophilia cases, compared to about 80% for hemophilia A [4].
- Severity tracks FIX activity ▾: severe (under 1%, frequent spontaneous bleeds), moderate (1–5%, bleeds after minor trauma), and mild (over 5–40%, bleeds only after major trauma or surgery).
- Clinical signs and symptoms ▾: The clinical hallmark is hemarthrosis, bleeding into joints such as knees, elbows, and ankles, which is clinically almost identical to hemophilia A.
- Diagnosis ▾: Prolonged aPTT with normal PT, confirmed by a low Factor IX activity assay, and distinguished from hemophilia A by a specific FIX assay.
- Treatment of hemophilia B ▾: Relies on Factor IX replacement and prophylaxis, and two gene therapies are now approved for eligible adults [7,8].
*Click ▾ for more information
Introduction
Hemophilia B is a lifelong inherited bleeding disorder caused by a shortage of Factor IX (FIX), a protein the body needs to form a firm blood clot. It is often introduced as the "other" hemophilia, overshadowed by the more common hemophilia A, but the two conditions look almost identical at the bedside. The difference lies one step earlier in the clotting chain, and that single difference shapes diagnosis, treatment choices, and even the strategy for managing rare complications.
This guide walks through hemophilia B from its molecular cause to its modern treatment.
Epidemiology
Hemophilia B affects approximately 1 in 25,000 to 30,000 male births, making it roughly one-fifth as common as hemophilia A [4]. It accounts for about 15% to 20% of all hemophilia cases, with hemophilia A making up most of the remainder [4]. Like hemophilia A, hemophilia B is X-linked and overwhelmingly affects males, though prevalence appears to vary somewhat by region and by how well local registries capture cases [4].
A note on women and girls
As with hemophilia A, heterozygous female carriers of an F9 mutation can have excessive bleeding due to skewed X-chromosome inactivation, and rare homozygous or compound heterozygous females have been reported [4]. These women may have hemophilia B in their own right rather than simply "carrying" it silently, and unexplained bleeding in a female relative of an affected male is worth investigating rather than dismissing.
Historical context: Christmas disease
Hemophilia B earned its older name, Christmas disease, from Stephen Christmas, the first patient in whom the condition was distinguished from hemophilia A, in 1952. Before that distinction, hemophilia A and B were treated as a single disorder, since the two are clinically almost impossible to tell apart without laboratory testing. The name is a helpful reminder of just how similar the two conditions are on the surface, and how much the underlying biology still matters.
Pathogenesis of Hemophilia B
This section explains why a missing protein one step earlier in the clotting chain produces a bleeding disorder that looks so much like hemophilia A, yet is managed somewhat differently.
The role of Factor IX
Factor IX is a protein made in the liver that circulates in the blood in an inactive form. Unlike Factor VIII, which is a cofactor, Factor IX is itself an enzyme, specifically a serine protease. This is the key mechanistic difference between the two hemophilias: FVIII helps another enzyme work faster, while FIX is the enzyme doing the cutting.
When an injury triggers clotting, Factor IX is activated into FIXa, either by Factor XIa or by the tissue factor–Factor VIIa complex. FIXa alone is a fairly weak enzyme. Its full power only appears once it partners with its cofactor, Factor VIIIa, which is why a shortage of either FVIII or FIX cripples the same step of the clotting chain.
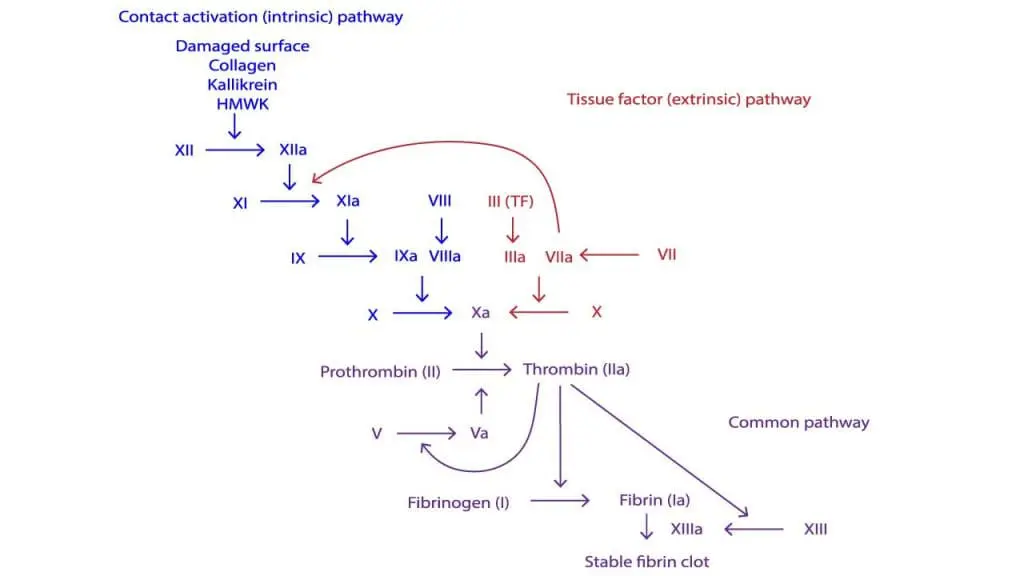
The tenase complex: the clotting assembly line
FIXa's job is to help build the intrinsic tenase complex, the same assembly line described in hemophilia A, but here FIXa is the engine rather than the cofactor. The complex has four parts, assembled on the surface of activated platelets: the enzyme (FIXa), the substrate (Factor X), the cofactor (FVIIIa), and calcium ions.
Once assembled, this complex activates Factor X at high speed, setting off the "thrombin burst" that builds a strong, stable fibrin clot. Without enough FIXa, the complex cannot form properly, no matter how much FVIII is present.
What goes wrong
In hemophilia B, low or absent FIX means the intrinsic tenase complex cannot assemble. Factor X activation slows to a crawl, and the thrombin burst needed for a firm clot never arrives. As in hemophilia A, the initial platelet plug still forms, since primary hemostasis does not depend on FIX, but the fibrin mesh reinforcing it is too weak and too slow to withstand pressure inside a joint or muscle. The clinical result, deep and delayed bleeding, is essentially identical to hemophilia A, even though the broken part of the machine is different.
The genetics behind it
Hemophilia B comes from mutations in the F9 gene, located near the end of the long arm of the X chromosome, at locus Xq27. More than 1,000 pathogenic variants have been described, most commonly missense and frameshift changes [4]. As in hemophilia A, the type of mutation broadly predicts severity.
- Missense mutations are the most common variant type and often allow some residual FIX function, producing a range of severities depending on exactly how the protein is affected.
- Null mutations, including large deletions, tend to produce severe disease with a complete absence of FIX protein, and carry a higher risk of inhibitor development, since the immune system has never encountered any FIX to recognize as "self" [4].
- A rare variant, Hemophilia B Leyden, causes low FIX activity in childhood that improves substantially after puberty, due to a promoter mutation that responds to androgens. This is a useful teaching example of how a single gene defect can have an age-dependent clinical course.
Clinical Signs and Symptoms
The clinical picture in hemophilia B is driven by the same principle as hemophilia A: less functional factor means more frequent, more severe, and more spontaneous bleeding. In fact, hemophilia A and B are considered clinically indistinguishable without laboratory testing.

Severity at a glance
The main types of bleeding
Hemarthrosis (joint bleeds). As in hemophilia A, this is the hallmark of the disease and the leading cause of long-term disability. Bleeding fills the joint space, most often in the knees, elbows, and ankles. Repeated bleeds trigger chronic joint inflammation that gradually destroys cartilage and bone, a process that ends in hemophilic arthropathy.
Intramuscular hematomas. Deep muscle bleeds can compress nerves or trigger compartment syndrome, both of which need urgent factor replacement.
Mucosal bleeding. Gum bleeding, prolonged nosebleeds, and occasional gastrointestinal bleeding can occur, particularly in more severe disease.
Life-threatening bleeds. Intracranial hemorrhage remains the leading cause of hemophilia-related death and can occur spontaneously or after minor trauma. Bleeding into the throat or neck can obstruct the airway. Both require immediate, aggressive factor replacement.
In newborns with a family history, watch for bleeding under the scalp, intracranial bleeding after a difficult delivery, or heavy bleeding after circumcision performed before diagnosis.
Diagnosis and Monitoring
Diagnosis follows the same sequence used in hemophilia A: a bleeding history or family history prompts screening tests, followed by a specific factor assay to confirm the diagnosis and pin down which factor is missing.
First-line screening tests
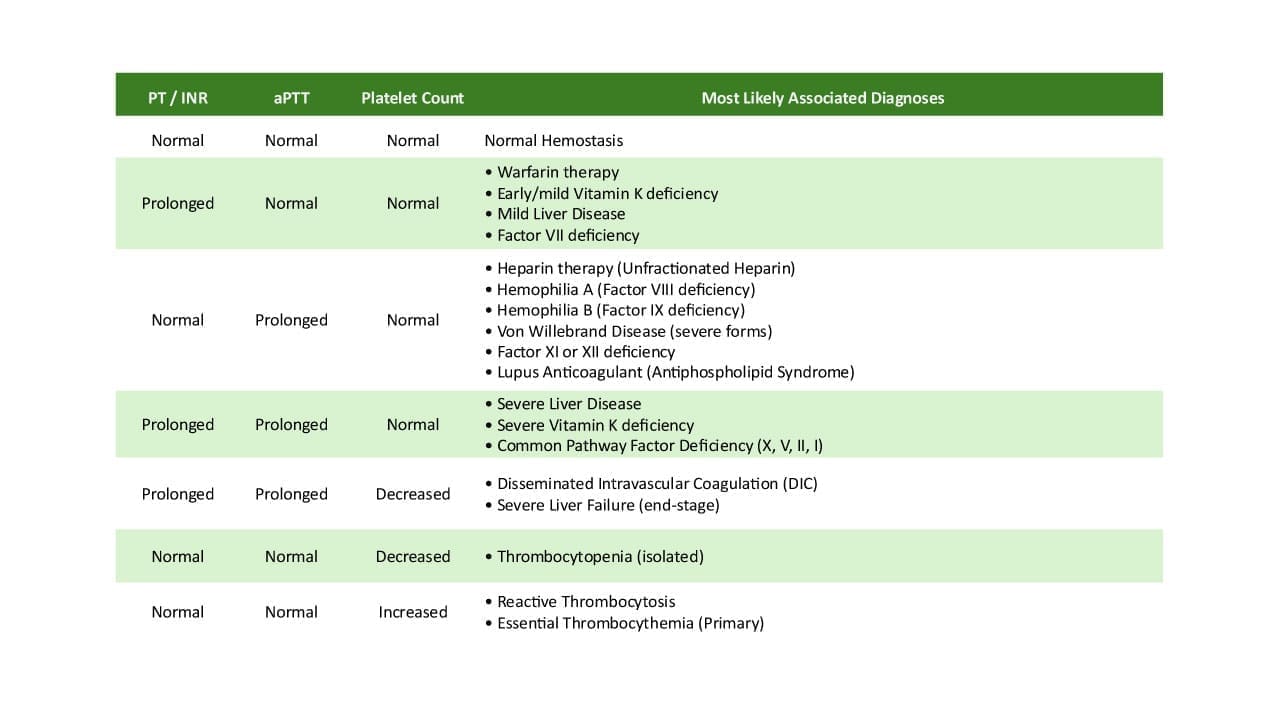
| Test | Result in hemophilia B | Why |
|---|---|---|
| aPTT | Prolonged | FIX is part of the intrinsic pathway, so its absence slows this test. |
| PT | Normal | The extrinsic and common pathways do not depend on FIX. |
| Thrombin time | Normal | The final clotting step is unaffected. |
| Platelet count | Normal | Hemophilia B is a clotting-protein disorder, not a platelet disorder. |
A prolonged aPTT with a normal PT looks identical whether the missing factor is VIII or IX. This is exactly why a specific factor assay is essential; screening tests alone cannot tell hemophilia A and B apart.
Confirming the diagnosis
The Factor IX activity assay (FIX:C) is the gold standard. It measures how well the patient's plasma corrects the clotting time of FIX-deficient plasma, and the result, expressed as a percentage of normal, sets the severity grade.
A Factor IX antigen level measures how much FIX protein is present, working or not. Most patients show low activity and low antigen together. A minority produce a normal amount of a non-functional protein, in which case antigen may be near-normal while activity is low, similar to the pattern occasionally seen in hemophilia A.
Laboratory Monitoring with New Therapies
Standard aPTT and specific FIX activity assays are often inaccurate or uninterpretable for measuring hemostatic potential in patients taking non-factor therapies like marstacimab or concizumab [11]. Once a patient transitions to these medications, traditional factor level monitoring cannot be used to assess treatment efficacy, and alternative specialized tests (such as thrombin generation assays) are required to evaluate the patient's coagulation status [11,12].
Genetic testing
Genetic testing identifies the specific F9 variant. This matters for two reasons: certain mutations, particularly large deletions and null mutations, carry a higher risk of inhibitor development [4], and knowing the exact variant allows accurate screening of at-risk family members.
Differential diagnosis
The key differential for a prolonged aPTT with a normal PT is hemophilia A, distinguished by a low Factor VIII level with a normal Factor IX level. Severe von Willebrand disease can also prolong the aPTT indirectly, but this is told apart by measuring von Willebrand factor levels, which are normal in hemophilia B.
Treatment and Management of Hemophilia B
The treatment goals mirror those in hemophilia A: prevent joint damage through prevention, and treat bleeds quickly when they occur. Care has moved from reactive treatment toward proactive prophylaxis, with major gains in quality of life as a result.
The Shift in Prophylaxis: Factor Replacement and Non-Factor Therapies
Historically, treatment centered entirely on raising FIX activity in the plasma via intravenous infusions. Recombinant Factor IX (rFIX) and extended half-life (EHL) products remain a standard approach, particularly for on-demand treatment or surgical coverage.
However, clinical management has undergone a major paradigm shift with the introduction of subcutaneous non-factor "rebalancing" therapies. These drugs do not replace Factor IX; instead, they block natural anticoagulants (like Tissue Factor Pathway Inhibitor, or TFPI) to allow the blood to clot even when Factor IX is missing.
- Marstacimab (Hympavzi): Approved by the FDA in late 2024 as a once-weekly subcutaneous injection, this tissue factor pathway inhibitor (TFPI) antagonist is indicated for routine prophylaxis in patients 12 years and older without inhibitors [9].
- Concizumab (Alhemo): Another anti-TFPI therapy, administered as a once-daily subcutaneous injection. It is approved for routine prophylaxis in patients 12 years and older, importantly including those with inhibitors [10].
These therapies have drastically reduced the treatment burden, moving many patients away from frequent intravenous infusions toward easier subcutaneous prophylaxis.
Treatment Strategies
Prophylaxis: Prevent bleeds by maintaining hemostasis. This is increasingly achieved using subcutaneous non-factor therapies (like marstacimab or concizumab) or via scheduled EHL FIX infusions.
On-demand: Treat a bleed as it happens, typically using intravenous rFIX.
Surgical cover: Provide high factor levels around a procedure, requiring intravenous FIX replacement before surgery and through healing.
Treating an acute bleed
Prompt FIX infusion is essential, especially for a joint bleed. As in hemophilia A, the R.I.C.E. approach (Rest, Ice, Compression, Elevation) supports factor infusion during an acute bleed.
When the immune system fights back: inhibitors
Inhibitors are less common in hemophilia B than in hemophilia A, developing in roughly 3% to 5% of patients with severe disease, though some cohort studies report figures as high as 9% to 23% [4,6]. This is meaningfully lower than the approximately 30% seen in severe hemophilia A.
However, inhibitors in hemophilia B carry a distinctive and serious risk that hemophilia A does not share: anaphylaxis. A meaningful proportion of patients who develop a FIX inhibitor also develop severe allergic or anaphylactic reactions to infused FIX, sometimes at the very first exposures [6].
Inhibitor warning
Unlike hemophilia A, where the first concern with an inhibitor is simply that treatment stops working, in hemophilia B an inhibitor can present as a medical emergency.
Immune Tolerance Induction (ITI) is used to eradicate inhibitors, as in hemophilia A, but it carries its own unique hazard in hemophilia B: a real risk of nephrotic syndrome, a kidney complication that can develop during high-dose FIX exposure [6]. ITI success rates in hemophilia B are also lower than in hemophilia A, in the range of 30% to 35% [6]. Because of the anaphylaxis risk, ITI in hemophilia B is sometimes preceded by gradual dose escalation, or desensitization, to reduce the chance of a severe reaction [6].
The Impact of Non-Factor Therapies on Inhibitor Management
The approval of anti-TFPI therapies like concizumab for patients with inhibitors has fundamentally altered how this complication is managed. Because these are non-factor therapies, they do not expose the patient to exogenous Factor IX. This effectively bypasses the risk of FIX-triggered anaphylaxis while providing steady-state prophylactic bleed protection [10]. As a result, the immediate necessity for high-risk Immune Tolerance Induction (ITI) and its associated risk of nephrotic syndrome has been significantly reduced for many patients.
Bypassing agents
When inhibitors are present and standard FIX replacement will not work, or carries an anaphylaxis risk, treatment shifts to agents that bypass the FVIII/FIX step entirely: activated prothrombin complex concentrate (aPCC) and recombinant Factor VIIa, both of which avoid re-exposing the patient to FIX itself.
Gene therapy: two approved options
Hemophilia B was the first hemophilia to receive an approved gene therapy, and two options now exist for eligible adults.
- Hemgenix (etranacogene dezaparvovec) received FDA approval in November 2022, becoming the world's first approved gene therapy for hemophilia B [7]. It uses an AAV5 vector carrying a high-activity variant of the F9 gene, called the Padua variant, which produces FIX with several times the normal specific activity. In clinical trials, the annualized bleeding rate fell by 54% compared to baseline [7].
- Beqvez (fidanacogene elaparvovec) received FDA approval in April 2024, becoming the second AAV-based gene therapy for hemophilia B [8]. It uses a different viral capsid (AAVRh74var) and is approved for adults with moderate to severe hemophilia B who are on prophylaxis or have a history of serious bleeding [8]. In its trial, patients achieved a median of zero bleeds over up to three years of follow-up [8].
Both therapies deliver a working F9 gene to liver cells via a one-time infusion, aiming to provide sustained, endogenous FIX production and reduce or eliminate the need for routine prophylaxis. As with hemophilia A gene therapy, patients are screened for pre-existing antibodies to the specific viral vector used, since these would neutralize the treatment before it reaches the liver.
While scientifically groundbreaking, real-world adoption of gene therapy has been gradual. High upfront costs, eligibility constraints (such as pre-existing AAV antibodies), and the competing convenience and high efficacy of the new subcutaneous non-factor therapies have positioned gene therapy as one of several highly effective options, rather than the automatic default choice for all patients [13].
Long-Term Complications
The long-term complications of hemophilia B closely mirror those of hemophilia A, since both stem from the same underlying problem: recurrent bleeding into joints and tissues.
Hemophilic arthropathy
Just as in hemophilia A, repeated hemarthrosis triggers chronic synovial inflammation, which leads to cartilage and bone destruction, target joints, and eventually permanent joint damage. Management follows the same principles: prevention through prophylaxis, physiotherapy to preserve joint function, and surgical options such as synovectomy or joint replacement for advanced disease.
Other long-term issues
- Bone health: reduced mobility and chronic inflammation can lower bone density over time.
- IV access devices: patients on long-term prophylaxis may need an implanted port, carrying infection and clotting risks.
- Historical viral infections: patients treated with older plasma-derived FIX products may carry chronic hepatitis C or HIV; this risk has been essentially eliminated with modern recombinant products.
- Psychological burden: the ongoing demands of infusions, chronic pain, and the fear of bleeding affect both patients and families.
Frequently Asked Questions
What is the main difference between hemophilia A and hemophilia B?
Hemophilia A is caused by a shortage of Factor VIII, a cofactor. Hemophilia B is caused by a shortage of Factor IX, an enzyme. Both sit in the same part of the clotting chain and produce nearly identical symptoms, but they are different genes, different proteins, and require different factor products for treatment.
Is hemophilia B more or less common than hemophilia A?
Less common. Hemophilia B makes up about 15% to 20% of hemophilia cases, compared to roughly 80% for hemophilia A, making hemophilia B about one-fifth as frequent [4].
Why is an inhibitor more dangerous in hemophilia B than in hemophilia A?
In hemophilia B, a FIX inhibitor can come with a real risk of anaphylaxis, a severe allergic reaction to infused FIX, sometimes appearing at the very first exposures. Treating an inhibitor with more FIX-based immune tolerance therapy also carries a distinctive risk of nephrotic syndrome, a kidney complication rarely seen in hemophilia A management [6].
Can hemophilia B be cured?
Not universally, but gene therapy is the closest option available. Two gene therapies, Hemgenix and Beqvez, are approved for eligible adults and deliver a working F9 gene so the liver can make its own Factor IX, reducing or removing the need for routine infusions in many patients [7,8].
What is the target trough Factor IX level for someone on prophylaxis?
The old benchmark was a trough above 1%. Current guidance favors individualized targets, since fewer bleeds generally occur at higher troughs, similar to modern practice in hemophilia A [2].
What is the most dangerous complication of hemophilia B?
Long term, it is hemophilic arthropathy, permanent joint damage from repeated bleeds. The most immediately dangerous single event is intracranial hemorrhage, which requires emergency factor replacement.
Glossary of Medical Terms
- Hemophilia B: An inherited bleeding disorder caused by a missing or faulty clotting protein called Factor IX. Also known historically as Christmas disease.
- Factor IX (FIX): A clotting enzyme made in the liver. Unlike Factor VIII, it is an active enzyme, not just a helper protein.
- Serine protease: A type of enzyme that cuts other proteins at specific points; Factor IX is one example.
- Tenase complex: The clotting "assembly line" where activated Factor IX, working with Factor VIIIa, activates Factor X.
- Hemarthrosis: Bleeding into a joint, the hallmark bleeding pattern in both hemophilia A and B.
- Hemophilic arthropathy: Long-term joint damage caused by repeated joint bleeds.
- Prophylaxis: Regular, scheduled treatment given to prevent bleeds before they happen.
- Trough level: The lowest level of clotting factor in the blood, measured just before the next dose is due.
- Inhibitors: Antibodies the immune system makes that attack infused Factor IX, making standard treatment stop working, and in hemophilia B, sometimes triggering allergic reactions.
- Anaphylaxis: A sudden, severe, potentially life-threatening allergic reaction; a distinctive risk when a hemophilia B inhibitor is present.
- Nephrotic syndrome: A kidney condition marked by protein loss in the urine; a known complication of immune tolerance therapy in hemophilia B.
- Immune Tolerance Induction (ITI): A treatment that gives frequent doses of factor concentrate to retrain the immune system to accept it.
- Gene therapy: A one-time treatment that delivers a working copy of the F9 gene so the liver can make its own Factor IX.
- AAV (adeno-associated virus): The modified, harmless virus used to deliver the therapeutic gene into liver cells.
- aPTT (activated partial thromboplastin time): A clotting screening test that is prolonged in both hemophilia A and B.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl 6): 1-158. https://doi.org/10.1111/hae.14046
- https://www.rarediseaseadvisor.com/disease-info-pages/hemophilia-guideline-recommendations/
- Berntorp, E., Fischer, K., Hart, D. P., Mancuso, M. E., Stephensen, D., Shapiro, A. D., & Blanchette, V. (2021). Haemophilia. Nature reviews. Disease primers, 7(1), 45. https://doi.org/10.1038/s41572-021-00278-x
- Konkle BA, Nakaya Fletcher S. Hemophilia B. 2000 Oct 2 [Updated 2025 Aug 7]. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1495/
- Castaman, G., & Matino, D. (2019). Hemophilia A and B: molecular and clinical similarities and differences. Haematologica, 104(9), 1702–1709. https://doi.org/10.3324/haematol.2019.221093
- Batorova, A., Morongova, A., Tagariello, G., Jankovicova, D., Prigancova, T., & Horakova, J. (2013). Challenges in the management of hemophilia B with inhibitor. Seminars in thrombosis and hemostasis, 39(7), 767–771. https://doi.org/10.1055/s-0033-1356574
- U.S. Food and Drug Administration / CSL Behring. (2022). FDA approval of HEMGENIX (etranacogene dezaparvovec-drlb), BLA 125772, for adults with Hemophilia B. https://www.fda.gov/vaccines-blood-biologics/vaccines/hemgenix
- Pfizer Inc. (2024). U.S. FDA Approves Pfizer's BEQVEZ (fidanacogene elaparvovec-dzkt), a one-time gene therapy for adults with hemophilia B. https://www.pfizer.com/news/press-release/press-release-detail/us-fda-approves-pfizers-beqveztm-fidanacogene-elaparvovec
- U.S. Food and Drug Administration. (2026). FDA expands approval of HYMPAVZI (marstacimab-hncq) for Hemophilia A and B. https://www.pfizer.com/news/press-release/press-release-detail/us-fda-approves-pfizers-hympavzi-treatment-two-additional
- https://www.bleeding.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-293-recommendation-on-the-use-and-management-of-anti-tissue-factor-pathway-inhibitors-in-hemophilia
- Lenting P. J. (2020). Laboratory monitoring of hemophilia A treatments: new challenges. Blood advances, 4(9), 2111–2118. https://doi.org/10.1182/bloodadvances.2019000849
- Kitchen, S., Tiefenbacher, S., & Gosselin, R. (2017). Factor Activity Assays for Monitoring Extended Half-Life FVIII and Factor IX Replacement Therapies. Seminars in thrombosis and hemostasis, 43(3), 331–337. https://doi.org/10.1055/s-0037-1598058
- Glasner, M. F., & Miesbach, W. (2025). Gene therapy for hemophilia: results of ISTH global survey on current knowledge, attitudes, and preparedness of the hemophilia care team. Therapeutic advances in hematology, 16, 20406207251384802. https://doi.org/10.1177/20406207251384802